Болезнь Вильсона
*Пятилетний импакт фактор РИНЦ за 2020 г.
Читайте в новом номере
В 1912 г. С.А. Вильсон впервые описал несколько семейных случаев синдрома прогрессирующей лентикулярной дегенерации у больных циррозом печени. Патогенез этого заболевания был раскрыт в 1948 г., когда впервые появились данные Кумингса о повышенном уровне меди в печени и головном мозге пациентов с болезнью Вильсона (БВ). С этого времени достигнут значительный прогресс в понимании физиологической функции меди и механизмов ее гепатотоксичности, расшифрован генетический дефект БВ. Эффективная и своевременная лекарственная терапия, направленная на снижение содержания меди в организме, позволила отнести это заболевание к немногим метаболическим заболеваниям печени, которые поддаются лечению.
БВ представляет собой редкое наследственное заболевание с аутосомно-рецессивным типом наследования, проявляющееся преимущественно в молодом возрасте и характеризующееся избыточным накоплением меди в организме. На начальных стадиях болезни медь накапливается в печени, что приводит к развитию гепатита, цирроза печени (ЦП) или фульминантной печеночной недостаточности (ФПН). На следующих стадиях избыток меди попадает в другие органы и системы, вызывая их повреждение, и прежде всего в головной мозг, приводя к нервно-психическим изменениям.
Содержание меди в обычной диете составляет
2–5 мг в день. К продуктам с высоким содержанием меди относятся: необработанная пшеница, бобы, горох, фасоль, моллюски, шоколад, печень, почки. В желудочно-кишечном тракте медь активно транспортируется в эпителий проксимальной части тонкой кишки, где часть меди (40–75%) остается в клетке связанной со специфическим белком, экскретируется с фекалиями при десквамации эпителия. Другая часть – 25–60% – абсорбируется в систему воротной вены при участии специфического переносчика. Медь, связанная с белками и аминокислотами, транспортируется по воротной вене в печень, где остается ее значительная часть (около 90%). Лишь небольшая часть альбуминсвязанной меди ( 
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Болезнь Вильсона — Коновалова

Болезнь Вильсона — Коновалова (гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона) — редкое наследственное заболевание, нарушение метаболизма меди, характеризующееся ее избыточным накоплением в тканях и жизненно-важных органах (в основном в печени, почках, головном мозге, глазах).
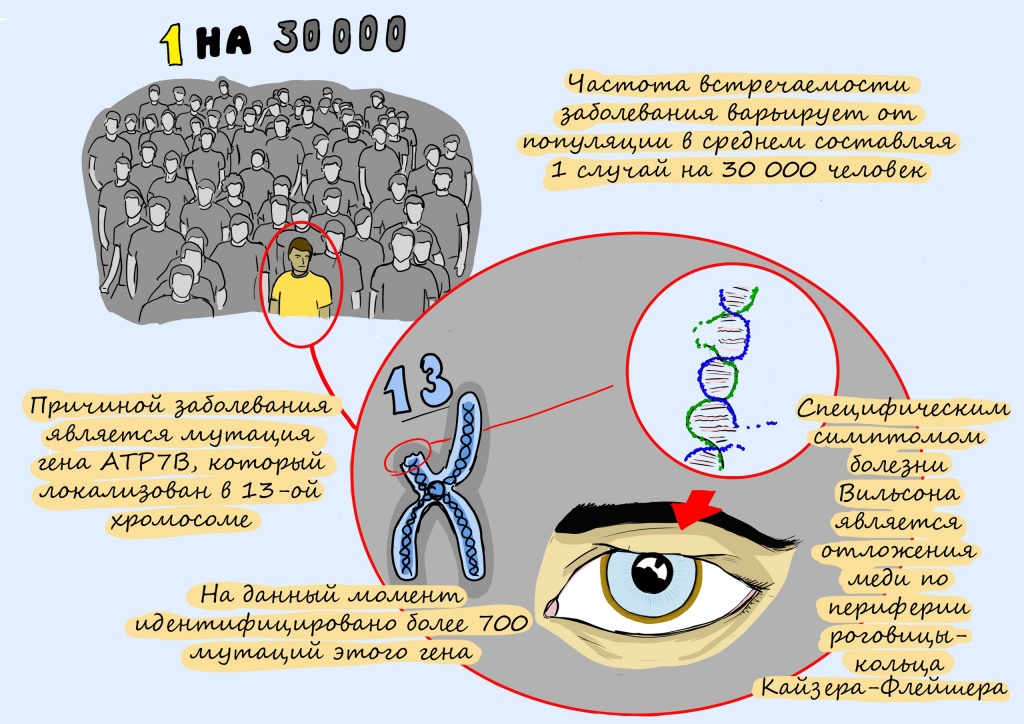
Распространенность болезни Вильсона — Коновалова во всем мире составляет 1 случай на 30 000–100 000 человек. Данные приблизительные, поскольку пациенты с гепатолентикулярной дегенерацией иногда получают ошибочный диагноз (в этот список входят различные болезни печени, неврологические заболевания и психические расстройства). Оценка носительства гена заболевания колеблется, однако считается, что его носителем может быть 1 человек из 90.
Причины возникновения
Болезнь Вильсона — Коновалова наследуется по аутосомно-рецессивному типу (мутации гена — в данном случае ATP7B — наследуются от обоих родителей). При таком типе наследования у каждого ребенка носителей дефектного гена существует 25% вероятность заболеть и 50% вероятность носительства мутации.
Ген ATP7B отвечает за метаболизм меди и играет важную роль в выведении избытка меди из организма. На сегодняшний день выявлено более 300 мутаций гена ATP7B.
Симптомы
Возраст манифестации заболевания варьируется: симптомы могут начать проявляться у ребенка 3–5 лет или же болезнь не дает о себе знать десятилетиями. Однако чаще всего болезнь Вильсона — Коновалова развивается к сорока годам, в редких случаях — после пятидесяти лет. Клиническая картина заболевания зависит от пола и возраста, так, у детей (средний возраст — 10 лет) чаще преобладают поражения печени.
К признакам и симптомам заболевания относятся:
Диагностика
Раннее выявление болезни Вильсона — Коновалова и своевременное лечение позволяют сохранить нормальное качество жизни пациента и предотвратить опасные осложнения. Диагноз устанавливается на основании физикального осмотра, сбора жалоб, личного и семейного анамнеза пациента, лабораторной диагностики (проводятся биохимические анализы крови и мочи, которые могут показать сниженную концентрацию медь-содержащего белка церулоплазмина в плазме крови, повышение уровня печеночных ферментов, тромбоцитопению, повышенное содержание меди в моче), осмотра глаз офтальмологом с помощью щелевой лампы, КТ или МРТ головного мозга (у пациентов с неврологическими симптомами). Если лабораторные исследования не подтверждают и не исключают диагноз, врач может назначить биопсию печени, а также молекулярно-генетическое тестирование.
Болезнь Вильсона — Коновалова дифференцируют (различают) со следующими заболеваниями: вирусный гепатит, аутоиммунный гепатит, неалкогольная жировая болезнь печени, алкогольный цирроз печени, первичный склерозирующий холангит, первичный билиарный цирроз, неврологическими патологиями (эссенциальный тремор, болезнь Паркинсона с ранним началом, дистония, болезнь Хантингтона).
Лечение болезни Вильсона — Коновалова
Во время беременности лечение заболевания рекомендуется не прекращать, в этот период врач может снизить дозировку хелатирующего агента, прием ацетата цинка продолжается по стандартной схеме. Кормление грудью при использовании хелатирующего агента исключается, данных по ацетату цинка недостаточно.
При острой печеночной недостаточности, вызванной прогрессированием болезни Вильсона — Коновалова, необходима трансплантация печени.
Особенности и преимущества лечения болезни Вильсона — Коновалова в клинике Рассвет
Врачи клиники Рассвет специализируются на диагностике и лечении редких синдромов и болезней, в область их интересов в том числе входит помощь пациентам с гепатолентикулярной дегенерацией (болезнью Вильсона — Коновалова).
Наши гепатологи — врачи высокой квалификации, прекрасно подготовлены и имеют большой практический опыт. Любое редкое заболевание требует мультидисциплинарного подхода, поэтому пациент с болезнью Вильсона — Коновалова при необходимости направляется на консультацию к офтальмологу, нефрологу, психотерапевту, психиатру.
Пациентам с подозрением на болезнь Вильсона — Коновалова проводятся все необходимые диагностические исследования для подтверждения или исключения диагноза, индивидуально подбирается схема терапии.
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФазы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Болезнь Вильсона может протекать в брюшной, ригидно-аритмогиперкинетической, дрожательной или экстрапирамидно-корковой форме. Диагностика болезни Вильсона включает офтальмологическое обследование, биохимические анализы мочи и крови, МРТ или КТ головного мозга. Основу патогенетической терапии составляют тиоловые препараты, которые могут приниматься в течении нескольких лет и даже пожизненно.
МКБ-10
Общие сведения
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФ-азы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Первооткрыватель заболевания — А.К. Вильсон, описавший заболевание в 1912 году, в отечественной медицине — Н.А. Коновалов. Патогенез болезни Вильсона был выявлен в 1993 году. Понятию «болезнь Вильсона» соответствуют также: болезнь Вильсона-Коновалова, болезнь Вестфаля-Вильсона-Коновалова, дистрофия гепатоцеребральная, дистрофия гепатолентикулярная, дегенерация лентикулярная прогрессирующая.
Причины
Ген АТР7В картирован на длинном плече хромосомы 13 (13q14.3-q21.1). Организм человека содержит около 50-100 мг меди. Суточная потребность меди для человека — 1-2 мг. 95% абсорбированной в кишечнике меди, транспортируется в форме комплекса с церулоплазмином (один из глобулинов сыворотки, синтезируемых печенью) и только 5% в форме комплекса с альбумином. Кроме того, ион меди входит в состав важнейших метаболических ферментов (лизилоксидаза, супероксиддисмутаза, цитохром-С-оксидаза и др.). При болезни Вильсона происходит нарушение двух процессов обмена меди в печени — биосинтез главного медьсвязывающего белка (церулоплазмина) и выведение меди с желчью, следствием чего становится повышение уровня несвязанной меди в крови. Концентрация меди в различных органах (чаще всего в печени, почках, роговице и головном мозге) увеличивается, что приводит к их токсическому поражению.
Классификация
Согласно классификации Н.В. Коновалова различают пять форм болезни Вильсона:
Симптомы
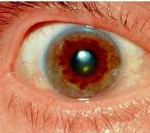
Для болезни Вильсона характерен клинический полиморфизм. Первые проявления заболевания могут появиться в детстве, юношестве, в зрелом возрасте и гораздо реже в зрелом возрасте. В 40-50% случаев Болезнь Вильсона манифестирует с поражения печени, в остальных — с психических и неврологических расстройств. С вовлечением в патологический процесс нервной системы обнаруживается кольцо Кайзера-Флейшера.
Брюшная форма развивается преимущественно до 40 лет. Характерный признак — тяжелое поражение печени по типу цирроза печени, хронического гепатита, фульминантного гепатита.
Ригидно-аритмогиперкинетическая форма манифестирует в детском возрасте. Начальные проявления — мышечная ригидность, амимия, смазанность речи, трудности при выполнении мелких движений, умеренное снижение интеллекта. Для этой формы заболевания характерно прогрессирующее течение, с наличием эпизодов обострения и ремиссии.
Дрожательная форма возникает в возрасте от 10 до 30 лет. Преобладающим симптомом является тремор. Кроме того, могут наблюдаться брадикинезия, брадилалия, тяжелый психоорганический синдром, эпилептические приступы.
Экстрапирамидно-корковая форма встречается весьма редко. Ее начало схоже с началом какой-либо из вышеперечисленных форм. Для нее характерны эпилептические припадки, экстрапирамидные и пирамидные нарушения и выраженный интеллектуальный дефицит.
Диагностика
Офтальмологическое исследование с помощью щелевой лампы выявляет кольцо Кайзера-Флейшера. Биохимические исследования мочи обнаруживают повышенную экскрецию меди в суточной моче, а также снижение концентрации церулоплазмина в крови. С помощью визуализационных методов (КТ и МРТ головного мозга) обнаруживают атрофию полушарий большого мозга и мозжечка, а также базальных ядер.
При диагностике болезни Вильсона неврологу необходимо дифференцировать ее от паркинсонизма, гепатоцеребрального синдрома, болезни Геллервордена-Шпатца. Основным дифференциально-диагностическим признаком этих заболеваний является отсутствие характерных для болезни Вильсона кольца Кайзера-Флейшера и расстройств обмена меди. Для подтверждения болезни Вильсона проводится генодиагностика.
Лечение болезни Вильсона
Основой патогенетического лечения является назначение тиоловых препаратов, в первую очередь — D-пеницилламина либо унитиола. Главное преимущество купренила — низкая токсичность и возможность длительного приема при отсутствии побочных эффектов. Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза). Лечение D-пеницилламином проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов). В случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности — леводопы, карбидопы, тригексифенидила.
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается обмен меди в организме. В дальнейшем лечение пациента составляет иммуносупрессивная терапия. В России на сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми клетками селезенки и печени (т. н. аппарат «вспомогательная печень). Немедикаментозное лечение состоит в назначении диеты (стол №5) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т. д.).
Прогноз и профилактика
В случае своевременного диагностирования болезни Вильсона и проведения адекватной медьснижающей терапии возможна нормализация общего состояние пациента и обмена меди в организме. Постоянный прием тиоловых препаратов по схеме, назначенной врачом-специалистом, позволяет поддерживать профессиональную и социальную активность пациента.
Для предотвращения рецидивов болезни Вильсона рекомендовано проведение лабораторных исследований крови и мочи пациента несколько раз в год. Необходим контроль следующих показателей: концентрация меди, церулоплазмина и цинка. Кроме того, рекомендовано проведение биохимического анализа крови, общего анализа крови, а также регулярные консультации у терапевта и невролога.
Медные люди: что важно знать о болезни Вильсона-Коновалова
Откуда берется медь в нашем организме и зачем она нужна? Из-за чего возникает болезнь, при которой медь не выводится из тела самостоятельно, и почему она опасна? Как живут люди, у которых меди слишком много, и чем медицина может им помочь?
Вместе с информационно-просветительским гуманитарным проектом «12 месяцев» мы запускаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними. Читайте в ноябре рассказ о болезни Вильсона-Коновалова, которая встречается у одного из 30 тысяч человек, а также историю «медного пациента» — 38-летней Олеси Фрерихс, которая узнала о заболевании на 7 месяце беременности.
Как связаны медь, гены и болезнь Вильсона-Коновалова?
Ежедневно наш организм получает с пищей не только белки, жиры, углеводы и витамины — в него поступает множество ионов металлов и других микроэлементов, необходимых для его правильного функционирования. Например, медь, которая содержится в печени и мясе, какао и бобовых, злаках и орехах.
Несмотря на небольшую суточную потребность — всего 1,5-2,5 мг, медь участвует в обмене энергии, метаболизме железа, защите клеточных мембран — то есть практически во всех физиологических процессах в нашем теле.
Обмен меди, как и многие другие индивидуальные особенности организма генетически запрограммированы, то есть заложены в нас при рождении. Гены — это всего лишь инструкции для синтеза белков в рибосомах. Один ген — один белок, все достаточно просто. А вот какую функцию будет выполнять этот белок — зависит от его структуры.
Некоторые белки участвуют в метаболизме меди. У всех людей всасывание меди происходит в желудке и двенадцатиперстной кишке. Дальше медь транспортируется в печень, где соединяется с различными белками, затем ее часть выводится в связанном состоянии в кровь и уже после — в мочу и кал.
Чтобы меди в организме было всегда ровно столько, сколько нужно, метаболизм регулирует и выравнивает скорости ее поступления и выведения наружу.
Ключевую роль в этом уравнении играет белок-транспортер меди под названием ATP7. Он работает исправно, если в его гене, в инструкции по его сборке, нет опечаток — или мутаций, как их называют биологи. Известно уже более 800 таких опечаток в гене АТР7.
Именно из-за этих мутаций у некоторых людей белок ATP7 не работает вовсе или его функция заметно снижена. В этом случае излишки меди не выводятся из организма, а накапливаются в органах. Но много — не всегда значит хорошо. Избыток металла не дает пациентам с неработающим белком ATP7 повышенную крепкость организма, а напротив — повреждает клеточные структуры. От избытка меди прежде всего страдает головной мозг и печень.
Такую болезнь, вызванную накоплением меди в организме из-за мутации белка ATP7, называют болезнью Вильсона — по имени Сэмюэля Уилсона (Вильсона) — британского невролога, подробно описавшего симптомы заболевания в 1912 году.
В России более распространено другое название — болезнь Вильсона-Коновалова: в 1960 году советский невропатолог Николай Коновалов существенно расширил понимание болезни. Еще реже можно встретить тройное название — Вильсона-Вестфаля-Коновалова (немецкий патолог Карл Фридрих Вестфаль описал болезнь еще в 1883 году). Сами пациенты называют себя вильсонятами.
В этом ролике — краткая история открытий на пути к познанию природы этой болезни и способов помочь пациентам.
Когда появляются симптомы и какими они могут быть?
Первые симптомы болезни Вильсона-Коновалова обычно появляются на втором или третьем десятке лет жизни в виде неврологических нарушений, например, нечеткости речи, нарушении глотания, автоматических жевательных движений, и нарушений показателей работы печени.
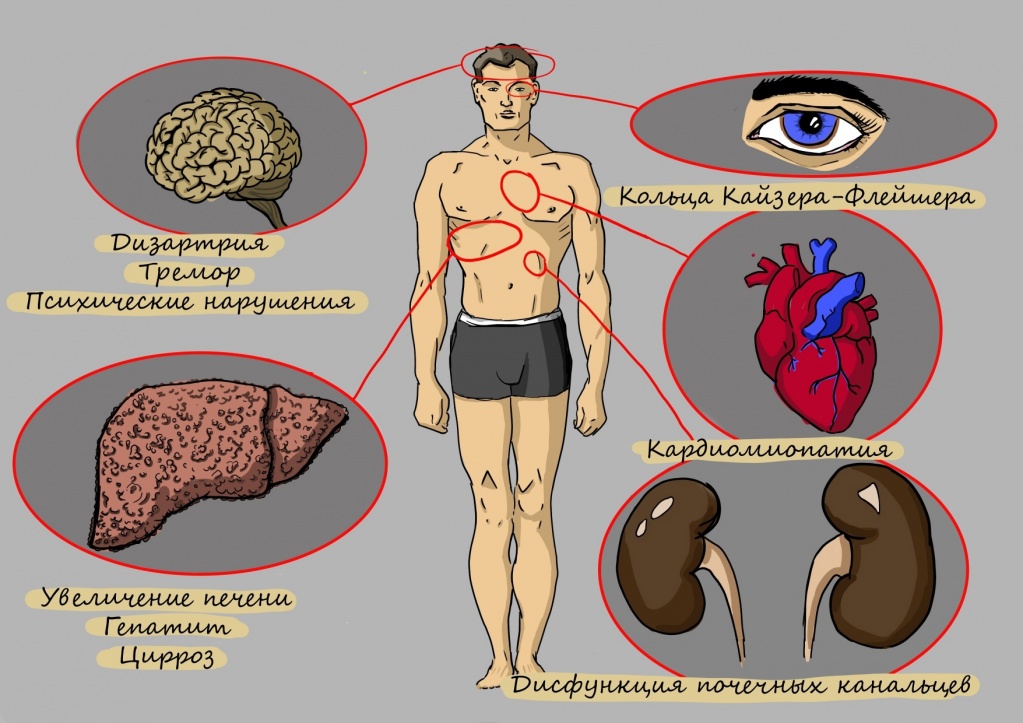
В теле не остается ни одного органа, равнодушного к нарушению обмена меди.
Чем медицина может помочь «медным людям»?
Главный на сегодняшний день способ лечения болезни Вильсона-Коновалова был предложен еще больше полувека назад — это регулярное применение препаратов, которые связывают медь и выводят ее из организма. Важным для «вильсонят» остается соблюдение строгой диеты и, при необходимости, пересадка печени.
Прерывание терапии или неправильное лечение может привести к смерти в течение нескольких месяцев. При этом медикаментозная, лекарственная терапия эффективна не для всех пациентов — побочные эффекты препаратов могут даже утяжелять состояние некоторых людей. Поэтому наука продолжает искать ответ — чем же помочь «вильсонятам»?
Не так давно исследователи научились исправлять «опечатки» в генах. В том числе — и в ATP7. Не всегда это получается хорошо, тем не менее, генная терапия может навсегда избавить пациентов с болезнью Вильсона-Коновалова от пожизненного приема препаратов и строгой диеты.
Как исследователи пытались научиться делать такую «операцию» на генах? Первые способы замены поврежденного участка гена на здоровый, в частности, применение олигонуклеотидов — коротких фрагментов ДНК или РНК, получаемых путем химического синтеза, даже у мышей приводили к едва заметному улучшению — его точно не хватило бы для помощи человеку.
Разочарования исследователей продлились до тех пор, пока они не научились «протезировать» больной ген. Для этого копию гена без поломки помещают в структуру искусственного вируса, чтобы эта неизмененная копия гена легко проникла в клетку. Освободившись от белковой оболочки, как от скафандра, ген начинает работать — и клетка наполняется правильно работающими переносчиками для меди.
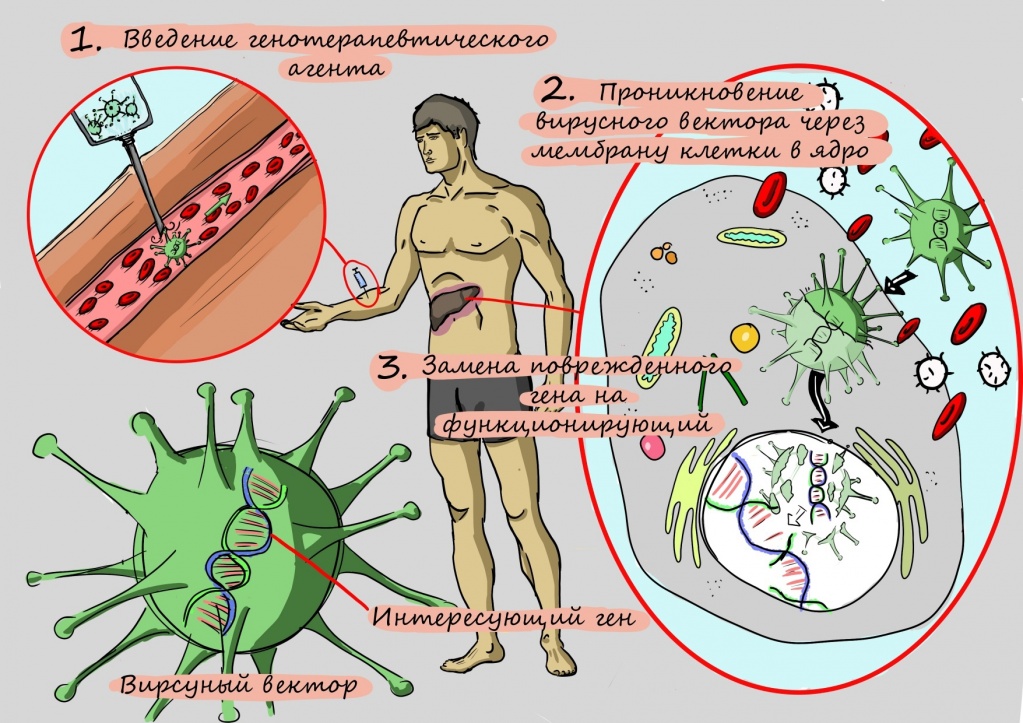
Молекулярное протезирование гена АТР7 — генная терапия
Однако при проведении этого вида терапии исследователи столкнулись с проблемой: аденовирус, который использовали для доставки гена в клетку, не может внедряться в собственный генетический аппарат клеток, — и это приводило к тому, что терапия работала недолго.
Следующим шагом в разработке терапии стало применение другого носителя — так называемого аденоассоциированного вирусного вектора.
Мышам с мутацией, которая похожа на мутацию в белке ATP7, вызывающую болезнь Вильсона-Коновалова, вводили такой вектор, и эффект оказался положительным.
Аденоассоциированный вирусный вектор имеет способность встраиваться в генетический аппарат клеток, поэтому результат лечения был более долгосрочным. Однако и этого оказалось недостаточно.
Успешный опыт подогревал ажиотаж исследователей: так, в 2021 году начались первые два клинических исследования по спасению «вильсонят» методами генной терапии (NCT04884815, NCT04537377). Через несколько лет мы узнаем — помогут ли эти разработки пациентам.
Материал подготовили: Роман Деев, Максим Пушкин, Екатерина Пичугина, Алексей Паевский, Виктория Рыжкова.
Иллюстрации Владислава Ефремова.
Болезнь Вильсона-Коновалова у детей
Общая информация
Краткое описание
Одобрен объединенной комиссией по качеству медицинских услуг
Министерства здравоохранения Республики Казахстан
от «28» ноября 2017 года
Протокол №33
Болезнь Вильсона-Коновалова (синонимы гепатолентикулярная дегенерация, гепатоцеребральная дистрофия) – тяжелое прогрессирующее наследственное заболевание, передающееся по аутосомно-рецессивному типу, в основе которого лежит нарушение экскреции меди из организма, приводящее к избыточному накоплению этого микроэлемента в тканях и сочетанному поражению паренхиматозных органов (прежде всего печени) и головного мозга (преимущественно подкорковых ядер).
NB! Причиной возникновения БВК являются мутации гена ATP7B, который локализован на 13 хромосоме в локусе 13q14.3 и кодирует медь транспортирующую АТФ-азу Р-типа – ATP7B.
Код(ы) МКБ-10:
| МКБ-10 | |
| Код | Название |
| E83.0 | Нарушение обмена меди (Болезнь Вильсона-Коновалова) |
Дата разработки/пересмотра протокола: 2017 год.
Сокращения, используемые в протоколе:
| анти-LC | антитела к цитозольному антигену печени |
| анти-LKM | антитела к микросомам печени и почек |
| анти-LP | антитела к белкам печени и поджелудочной железы |
| анти-SLA | антитела к растворимым печеночным антигенам |
| БВК | болезнь Вильсона-Коновалова |
| БХАК | биохимический анализ крови |
| ГГТП | Гамма-глютамилтранспептидаза |
| ГЛД | гепатолентикулярная дегенерация |
| КТ | компьютерная томография |
| МРТ | магниторезонансная томография |
| ОАК | общий анализ крови |
| ОАМ | общий анализ мочи |
| СОЭ | скорость оседания эритроцитов |
| СРБ | С-реактивный протеин |
| УЗИ | ультразвуковое исследование |
| ЩФ | Щелочная фосфотаза |
| ЭКГ | электрокардиограмма |
| ЭНМГ | электронейромиография |
| ЭРХПГ | Эндоскопическая ретроградная холангиопанкреатография |
| ЭЭГ | электроэнцефалография |
| ANA | антинуклеарные антитела |
| IgG | иммуноглобулин G |
| АNCА | антитела к цитоплазме нейтрофилов |
| АМА | антимитохондриальные антитела |
Пользователи протокола: врачи общей практики, педиатры, детские гастроэнтерологи, детские неврологи.
Категория пациентов: дети.
Шкала уровня доказательности:
| A | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| B | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| C | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+), результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GPP | Наилучшая клиническая практика. |

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
Клиническая картина гепатолентикулярной дегенерации характеризуется большим полиморфизмом в отношении как неврологических, так и соматических проявлений. Этот полиморфизм отражен в различных классификациях заболевания.
Формы болезни Вильсона [3]:
· Бессимптомная форма;
· Печеночная форма;
· Церебральная форма;
· Смешанная форма.
В зависимости от вовлечения в патологический процесс печени и центральной нервной системы и характера экстрапирамидной симптоматики, распознают 5 форм гепато-церебральной дистрофии [11]:
· Брюшная (абдоминальная) форма – манифестирует в возрасте от 5 до 17 лет и характеризуется различными вариантами поражения печени, нередко принимающими злокачественное «галопирующее» течение, приводящее к смерти раньше появления симптомов со стороны нервной системы. Её продолжительность от нескольких месяцев до 3-5 лет.
· Ригидно-аритмогиперкинетическая, или ранняя форма отличается быстрым течением; начинается также в детском возрасте. В клинической картине преобладают мышечная ригидность, приводящая к контрактурам, бедность и замедленность движений, хореоатетоидные или торсионные насильственные движения. Характерны дизартрия и дисфагия, судорожный смех и плач, аффективные расстройства и умеренное снижение интеллекта. Заболевание длится 2-3 года, заканчивается летально.
· Дрожательно-ригидная форма встречается чаще других; начинается в юношеском возраста, протекает медленнее, порой с ремиссиями и внезапными ухудшениями, сопровождающимися субфебрильной температурой; характеризуется одновременным развитием тяжѐлой ригидности и дрожания, дрожание очень ритмичное (2-8 дрожаний в секунду), резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает. Иногда обнаруживаются атетоидные хореоформные насильственные движения; наблюдаются также дисфагия и дизартрия. Средняя продолжительность жизни около шести лет.
· Дрожательная форма начинается в возрасте 20-30 лет, протекает довольно медленно (10-15 лет и больше); дрожание резко преобладает, ригидность появляется лишь в конце болезни, а порой наблюдается гипотония мышц; отмечается амимия, медленная монотонная речь, тяжѐлые изменения психики, часты аффективные вспышки. Наблюдаются эпилептиформные припадки.
· Экстрапирамидно-корковая форма встречается реже других форм. Типичные для гепатоцеребральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжѐлым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально.
Диагностика
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ [1-6,10,19,20]: на БВ должны обследоваться дети в возрасте от 2 до 18 лет, имеющих необъяснимое повышение сывороточных аминотрансфераз, проявления фульминантной печеночной недостаточности, хронического гепатита, цирроза печени, неврологические нарушения неустановленной этиологии, Кумбс-негативную гемолитическую анемию, отягощенный семейный анамнез по БВ. Диагностика БВ базируется на комбинации клинических симптомов, данных лабораторного обследования и молекулярно-генетического тестирования.
Диагностические критерии [1-4]
Жалобы:
· боли в животе различной локализации;
· изменение цвета кожи;
· носовые кровотечения;
· тремор и непроизвольные движения;
· слюнотечение, дизартрия, нарушение глотания;
· мигренеподобные головные боли;
· бессонница;
· депрессия;
· невротическое поведение;
· изменения личности;
· психоз.
Анамнез: Первичная манифестация БВ может протекать в виде острого фульминантного гепатита, проявляющегося коагулопатией, энцефалопатией, Кумбс-негативной гемолитической анемией, печеночноклеточной и почечной недостаточностью, с выявлением значительного превышения меди в сыворотке крови и моче.
NB! Обратить внимание на возраст начала проявлений заболевания у пациента: до 5 летнего возраста проявления болезни Вильсона-Коновалова, как правило, отсутствуют. Болезнь манифестирует в возрасте 8-16 лет (хотя практически с рождения отмечается повышенная активность печеночных аминотрансфераз). Необходимо уточнить о наличии заболеваний печени и нейропсихических нарушений у ближайших родственников больного (стеатоза, гепатитов, цирроза печени, печеночной недостаточности).
NB! Первые симптомы заболевания начинается с симптомов поражения печени (в 42% случаев). Примерно у 25% пациентов заболевание начинается остро, с развития желтухи, астенического синдрома, анорексии, повышения температуры. Болезнь Вильсона-Коновалова может клинически протекать по типу аутоиммунного гепатита, с повышением уровня сывороточных иммуноглобулинов и неспецифических аутоантител, в связи с чем необходимо исключать данное заболевание и у больных с аутоиммунным гепатитом. Неврологическая и психическая симптоматика наблюдается у 10% больных. У 15% пациентов болезнь Вильсона-Коновалова манифестирует гематологическими синдромами (прежде всего гемолитической анемией). Кольцо Кайзера-Флейшера не выявляется у детей до 5 лет.
| Проявления болезни Вильсона-Коновалова | Симптомы |
| Поражение печени | Бессимптомная гепатомегалия |
| Изолированная спленомегалия | |
| Стеатогепатит | |
| Острый (фульминантный) гепатит | |
| Аутоиммуноподобный гепатит | |
| Цирроз печени | |
| Поражение ЦНС | Двигательные нарушения (тремор, непроизвольные движения) |
| Слюнотечение, дизартрия | |
| Ригидная дистония | |
| Псевдобульбарный синдром | |
| Вегетососудистая дистония | |
| Мигренеподобные головные боли | |
| Бессоница | |
| Дистонические атаки | |
| Психиатрические симптомы | Депрессия |
| Невротическое поведение | |
| Изменения личности | |
| Психоз | |
| Другие системы | Офтальмология: кольца Кайзера-Флейшера, «медная» катаракта |
| Гемолитическая анемия | |
| Патология почек: аминоацидурия, нефролитиаз | |
| Патология скелета: ранний остеопороз, артрит | |
| Поражение сердца: кардиомиопатия, нарушения ритма | |
| Панкреатит, желчекаменная болезнь | |
| Гипопаратиреодизм, гигантизм |
Физикальное обследование 5:
необходимо оценить наличие:
· смуглого («медный») цвета кожи;
· желтушности склер;
· незначительной или умеренной гепатомегалии;
· спленомегалии;
· неврологических нарушений и психических расстройств в виде непроизвольных движений в мышцах торса и конечностей;
· мигренеподобной головной боли;
· скованности в мышцах;
· эмоциональной лабильности;
· агрессивности.
Лабораторные исследования 2:
· общий анализ крови: лейкопения, нормохромная анемия, тромбоцитопения, ретикулоцитоз, ускоренная СОЭ.
· общий анализ мочи: при поражении почек можно обнаружить микрогематурию, незначительную протеинурию, гиперкальциурию.
· суточная экскреция мочи: гиперкупренилурия, признаки развившейся тубулопатии с признаками: глюкозурией, аминоацидурией, фосфатурией, уратурией, протеинурией.
· биохимический анализ крови: снижение церулоплазмина и общей меди, увеличение уровней свободной меди (таблица 1), аминотрасфераз (в 1,5-50 раз); билирубин повышен более чем в 2 раза, преимущественно за счет прямой фракции; уровень щелочной фосфатазы обычно повышен; может быть повышена активность гаммаглютамилтранспептидазы (ГГТП); гипоальбуминемия.
· коагулограмма: снижение протромбинового индекса, гипофибриногенемия, снижение тромбинового времени.
· пеницилламиновый тест: необходимо исследовать мочу, собранную сразу после приема 500 мг пеницилламина и через 12 часов. У пациентов с болезнью Вильсона-Коновалова суточная экскреция меди будет повышаться до более 1500 мкг/дл/сут (норма
| Показатель | Норма | ГЛД |
| Церулоплазмин, мг/дл | 17–40 | |
| Общая медь в сыворотке крови, мкмоль/л | 12–32 | |
| Свободная медь в сыворотке крови, мкг/дл | 5–12 | > 50 |
| Суточная экскреция меди с мочой, мкг/сут | > 100–1000 | |
| Суточная экскреция меди с мочой в пробе с Д-пеницилламином, мкг/сут | 600–800 | 1000–3000 |
| Количественное содержание меди в печени, мкг/г | 15–55 | > 250 |
Показания для консультации специалистов 4:
· консультация офтальмолога – для выявления колец Кайзера-Флейшера, также на наличие катаракты у детей, с целью исключения других болезней накопления;
· консультация невропатолога – оценка неврологического статуса, нервно-психического статуса;
· консультация психиатра – диагностика психиатрических состояний;
· консультация психотерапевта – коррекция психологических проблем;
· консультация гастроэнтеролога – коррекция нарушений желудочно-кишечного тракта;
· консультация гематолога – при наличии симптомов гемолитической анемии, коагулопатии;
· консультация нефролога – при наличии патологии в анализах мочи;
· консультация сурдолога – определение остроты слуха;
· консультация физиотерапевта – определение методов физиотерапевтического лечения;
· консультация хирурга – при риске пищеводно-желудочных кровотечений, для выявления показаний к проведению трансплантации печени у детей с признаками цирроза печени (ЦП), печеночно-клеточной декомпенсацией;
· консультация инфекциониста – при наличии сопутствующего вирусного гепатита;
· консультация отоларинголога – при инфекциях верхних дыхательных путей.
Диагностический алгоритм: 
Рисунок 1- Алгоритм диагностики болезни Вильсона [3]. КФ – кольца Кайзера-Флейшера; Цер – церулоплазмин; 24-h Cu – суточная экскреция меди с мочой.
Ни один лабораторный тест (за исключением полного секвенирования патологического гена АТР7В) не обладает 100% чувствительностью и не обеспечивает 100% специфичность, таблица 3.
| Тест | Характерные находки | Ложноотрицательный результат | Ложноположительный результат |
| Сывороточный церулоплазмин | Уменьшение на 50% относительно нормы | Нормальный уровень у пациентов с выраженным воспалением в печени. Завышение результата при иммунологических методах исследования. Беременность, прием эстрогенов. | Низкий уровень при: -мальабсорбции; -ацерулоплазминемии; -гетерозиготы |
| Суточная экскреция меди с мочой | >0,64мкмоль/24часа | Норма: -неправильный сбор мочи; -дети без вовлечения печени | Повышение: -гепатоцеллюлярный некроз; -холестаз; -контаминация |
| Свободная медь сыворотки | >1,6мкмоль/л | Норма при завышении уровня церулоплазмина иммунологическими методами | |
| Печеночная медь | >4мкмоль/г сухого вещества | В зависимости от места взятия материала: -активная болезнь печени; -узелки регенерации | Синдромы холестаза |
| Кольцо Кайзера-Флейшнера (при использовании щелевой лампы) | Наличие кольца | Отсутствует: -у 50% пациентов с печеночной формой; -у большинства асимптоматических сибсов | Первичный биллиарный цирроз |
Диагноз болезни Вильсона ставится на основании совокупности клинических данных, результатов лабораторного исследования и молекулярно-генетического анализа (таблица 4).
| Типичные клинические симптомы и признаки | Другие тесты |
| Кольцо Кайзера-Флейшнера: Присутствует 2 Отсутствует 0 | Печеночная медь (при отсутствии холестаза) >5х верхней границы нормы (>4мкМ/г) 2 0,8-4мкМ/г 1 Норма ( |
| Неврологическая симптоматика или типичные изменения на МРТ головного мозга: Тяжелая 2 Средняя 1 Отсутствует 0 | Медь в моче (в отсутствии острого гепатита) Норма 0 1-2х верхней границы нормы 1 >2х верхней границы нормы 2 Норма, но увеличение >5х верхней границы нормы после пеницилламина 2 |
| Церулоплазмин сыворотки Норма (>0,2г/л) 0 0,1-0,2 г/л 1 | Анализ мутаций Обе хромосомы 4 На одной хромосоме 1 Не выявлено мутаций 0 |
| Кумбс-негативная гемолитическая анемия Присутствует 1 Отсутствует 0 | |
| Интерпретация результата | |
| 4 и более | Диагноз установлен |
| 3 | Диагноз возможен, необходимы дальнейшие исследования |
| 2 и менее | Диагноз маловероятен |
Дифференциальный диагноз
Дифференциальный диагноз и обоснование дополнительных исследований 17
Таблица 5– Дифференциальный диагноз болезни Вильсона-Коновалова.
Лечение
Препараты (действующие вещества), применяющиеся при лечении
| Клоназепам (Clonazepam) |
| Пантопразол (Pantoprazole) |
| Пеницилламин (Penicillamine) |
| Пиридоксин (Pyridoxine) |
| Ранитидин (Ranitidine) |
| Тиаприд (Tiapride) |
| Тригексифенидил (Trihexyphenidyl) |
| Триентин (Trientine) |
| Цинка ацетат (Zinc acetate) |
| Цинка оксид (Zinc oxide) |
| Цинка сульфат (Zinc sulfate) |
| Эзомепразол (Esomeprazole) |
Лечение (амбулатория)
ТАКТИКА ЛЕЧЕНИЯ НА АМБУЛАТОРНОМ УРОВНЕ [1-4,11]:
· поддерживающая хелатная терапия;
· диспансерное наблюдение.
Лечение (стационар)
ТАКТИКА ЛЕЧЕНИЯ НА СТАЦИОНАРНОМ УРОВНЕ [1-4,9-11]: все пациенты с данным диагнозом лечатся на стационарном уровне. Проводится подбор хелатной терапии с учетом индивидуальных особенностей пациента, подбор/коррекция хелатной терапии и подбор симптоматической терапии, м при необходимости ортотопическая трансплантация печени.
Карта наблюдения пациента, маршрутизация пациента:
| Продукт | Медь, мг в 100г | Продукт | Медь, мг в 100г |
| Печень телячья жареная | 23,9 | Малина | 0,170 |
| Печень баранья жареная | 13,5 | Абрикос | 0,170 |
| Устрицы | 7,5 | Редис | 0,150 |
| Угри вареные | 6,6 | Яйца куриные | 0,150 |
| Дрожжи сухие | 5,0 | Картофель | 0,140 |
| Какао-порошок | 3,9 | Свекла | 0,140 |
| Пюре томатное | 2,9 | Баклажан | 0,135 |
| Семена подсолнечника | 2,3 | Киви | 0,135 |
| Орехи кешью | 2,1 | Чеснок | 0,130 |
| Креветки вареные | 1,9 | Крыжовник | 0,130 |
| Крабы вареные | 1,8 | Смородиначерная | 0,130 |
| Орехи бразильские | 1,8 | Земляника садовая | 0,125 |
| Семена тыквенные | 1,6 | Салат | 0,120 |
| Семена кунжута | 1,5 | Груша | 0,120 |
| Тахини | 1,5 | Капуста брокколи | 0,120 |
| Омары вареные | 1,4 | Яблоки | 0,110 |
| Орехи грецкие | 1,3 | Помидор | 0,110 |
| Орехи кедровые | 1,3 | Редька | 0,100 |
| Фундук | 1,2 | Огурцы | 0,100 |
| Арахис | 1,0 | Перец красный сладкий | 0,100 |
| Кальмары | 1,0 | Вишня | 0,100 |
| Миндаль | 1,0 | Лук зеленый | 0,092 |
| Фисташки жареные соленые | 0,8 | Слива | 0,087 |
| Смородина | 0,8 | Лук репчатый | 0,085 |
| Горошек зеленый | 0,75 | Морковь | 0,080 |
| Арахисовое масло | 0,7 | Виноград | 0,080 |
| Грибы | 0,7 | Капуста зеленая | 0,075 |
| Чечевица | 0,66 | Сыр Чеддер | 0,070 |
| Греча ядрица | 0,64 | Сыр российский | 0,070 |
| рис | 0,560 | Сыр рассольный | 0,070 |
| геркулес | 0,450 | Апельсин | 0,067 |
| кукуруза | 0,290 | Сыр адыгейский | 0,060 |
| лимон | 0,240 | Дыня | 0,047 |
Медикаментозное лечение [10,11]:
Современная патогенетическая терапия ГЛД основана на использовании медьэлиминирующих препаратов [10,11], главным образом Д-пеницилламина, триентина и солей цинка. Д-пеницилламин и триентин – хелатные комплексоны, образующие с медью прочные соединения, которые легко выводятся из организма с мочой.
Препаратом выбора при ГЛД является Д-пеницилламин. Лечение начинают с небольшой дозы с постепенным увеличением ее до терапевтической, под контролем выделения меди с мочой. Начальные дозы составляют 250-500 мг/сут с постепенным (каждые 4-7 дней) увеличением дозы на 250 мг до лечебной дозировки 1000-1500 мг/сут, которая дается в 2-4 приема. Д-пеницилламин назначается за 1 час или через 2 часа после приема пищи, т.к. еда снижает кишечную абсорбцию препарата. Когда клиническое состояние пациента стабилизируется, а экскреция меди с мочой снизится, дозу препарата уменьшают. На фоне приема Д-пеницилламина у больных со смешанной (неврологической) формой БВ может отмечаться ухудшение неврологической симптоматики, обусловленное индуцированной высокой мобилизации меди из печени и отложением ее в базальных ядрах головного мозга, что провоцирует или усиливает неврологическую симптоматику. Побочные явления на фоне терапии Д-пеницилламином (лихорадка, кожная сыпь, тромбоцитопения, лейкопения и пр.) наблюдаются у 25% пациентов. Возможно развитие подострой токсической реакции в виде протеинурии, угнетения костномозгового кровообращения или хронического токсического действия на кожу (преждевременное старение, дефекты в формировании рубцовой ткани, серпингинозный перфорирующий эластоз, вследствие токсического воздействия на коллагеновые волокна, возможно развитие слабости сосудистой стенки), иммунную систему с развитием аутоиммунных заболеваний (системная красная волчанка, артриты, повышение антинуклеарного фактора), а также снижение резистентности к инфекциям. Прием препарата прекращают, после исчезновения побочных эффектов возобновляют с минимальной дозы.
Д-пеницилламин является антагонистом пиридоксина, поэтому к терапии следует добавить пиридоксин в дозе 25-50 мг в сутки.
При выраженной и сохраняющейся непереносимости Д-пеницилламина назначаются препараты цинка.
Цинк (оксид, сульфат, ацетат) индуцирует синтез медьсвязывающих белков (металлотионинов) в эпителии тонкой кишки и гепатоцитах, что препятствует абсорбции меди в портальную циркуляцию. При лечении цинком медь выводится через кишечник. Рекомендуемая доза – 25 мг 3 раза в день, у детей и беременных в 2 раза меньше. Недостаточный ответ на лечение сопровождается повышенной экскрецией меди с мочой (более 125 мкг/сут). Показаниями к назначению препаратов цинка являются: стойкий резидуальный неврологический синдром, остающийся на фоне многолетней терапии Д- пеницилламином; обострение неврологической и (или) печеночной симптоматики в начальной стадии терапии Д-пеницилламина; доклиническая и доневрологическая стадии ГЛД. Лечение также проводится пожизненно.
| Лекарственная группа | Лекарственные средства | Способ применения | Уровень доказательности |
| Хелатор меди | Д-пеницилламин | 20мг/кг/сут в 2-4 приема, через 2ч после приема пищи. Последняя таблетка через 3ч после ужина. | В |
| Триентин * | Дети 12 лет и взрослые: 750—1250 мг/сут в 2—4 приема; максимальная доза 2 г/сут. | В | |
| Витамины | Пиридоксина гидрохлорид | 10мг по 1таблетке 2 раза в день после еды | С |
* применение препарата возможно после регистрации на территории РК.
| Препарат | Неврологическое ухудшение | Побочные эффекты | Комментарии |
| Д-пеницилламин | В начале лечения в 10-20% | -лихорадка, сыпь, протеинурия, волчаночноподобные реакции -апластическая анемия -лейкопения -тромбоцитопения -нефротический синдром -дегенеративные изменения кожи -серозный ретинит -гепатотоксичность | Уменьшение дозы при операциях для улучшения заживления раны и во время беременности. Уменьшение дозы на 25% при ремиссии |
| Цинк 1 | Возможно при начале лечения | -гастрит; биохимический панкреатит -аккумуляция цинка -возможные изменения в иммунной системе | Нет необходимости уменьшения дозы при операции или во время беременности |
| Триентин | В начале лечения в 10-15% | -гастрит — редко апластическая анемия -сидеробластная анемия | Уменьшение дозы при операциях для улучшения заживления раны и во время беременности. Уменьшение дозы на 25% при ремиссии |
1 нет клинических данных о совместном применении цинка с хелатирующим агентом
Таблица 9 — Перечень дополнительных лекарственных средств (менее 100% вероятности применения):
| Лекарственная группа | Лекарственные средства | Способ применения | Уровень доказательности |
| Индуктор металлотинеинов, блокирует всасывание меди в кишечнике | Цинк | По 25мг х 3 раза в день за 30мин до еды | С |
| Ингибитор протонной помпы | Пантопразол | старше 12 лет назначают по 40-80 мг в день | В |
| Эзомепразол | старше 12 лет 40-80 мг 1-2 раз в сутки | В | |
| Н2-гистаминоблокатор | Ранитидин | старше 14 лет – 150 мг 2 раза в сутки или 300 мг на ночь | С |
| Антихолинергические средства | Тригексифенидила гидрохлорид | 5-15 мг/сут, максимально до 40 мг/сут; в 2-3 приема | В |
| Атипичный нейролептик | Тиаприд | 3-6 мг/кг/сут в 2-3 приема | В |
| Производное бензодиазепина | Клоназепам | от 1 года до 5 лет — 1,5—3 мг в сутки; от 6 до 16 лет — 3—6 мг в сутки, в 2- 3 приёма. | В |
Хирургическое вмешательство:
1) Трансплантация печени
Показания:
· развитие фульминантной печеночной недостаточности;
· неэффективность терапии хелаторами меди в течение нескольких месяцев у пациентов с декомпенсированным циррозом печени;
· возникновение тяжелой прогрессирующей печеночной недостаточности при самостоятельном прекращении лечения, прогрессирующих и необратимых неврологических нарушениях.
2) Склеротерапия, облитерация, лигирование, прошивание варикозных вен пищевода
Показания:
· варикозное кровотечение.
Дальнейшее ведение:
· диспансерное наблюдение пожизненное (в первый год – ежемесячно с контролем лабораторных показателей; далее – 4 раза в год);
· на амбулаторном этапе после выписки из стационара: профилактика интеркуррентных заболеваний, диета с низким содержанием меди, обычный режим, дозированные физические упражнения;
· длительно сохраняющиеся соматические симптомы не являются противопоказанием к посещению школы; при выраженных неврологических и психических изменениях – обучение в специализированных школах или индивидуальное.
Индикаторы эффективности лечения:
· нормализация функциональных проб печени;
· уменьшение/исчезновение симптомов заболевания.
NB! Прогноз и исходы болезни Вильсона/
Болезнь Вильсона является прогрессирующим заболеванием и при отсутствии своевременной терапии больные умирают от осложнений цирроза печени и/или реже от прогрессирующей неврологической симптоматики. При хелирующей терапии и трансплантации печени длительная выживаемость пациентов с болезнью Вильсона стала нормой, хотя и не оценивалась проспективно.
Прогноз при болезни Вильсона связан со степенью декомпенсации печеночных функций, тяжестью неврологической симптоматики и приверженностью терапии. Нормализация печеночных функций происходит на 1-2 году терапии и не прогрессирует при полном выполнении всех рекомендаций. Консервативная терапия не эффективна при фульминантном течении заболевания. Был разработан прогностический индекс болезни Вильсона (Dhawan et al.), согласно которому оценка свыше 11 баллов связана с высокой вероятностью летального исхода при отсутствии срочной ортотопической трансплантации печени, таблица 10.
| Показатель | 1 балл | 2 балла | 3 балла | 4 балла |
| Билирубин (мкмоль/л) | 100-150 | 151-200 | 201-300 | >300 |
| АСТ (Ед/л) | 100-150 | 151-300 | 301-400 | >400 |
| МНО | 1.3-1.6 | 1.7-1.9 | 2.0-2.4 | >2.4 |
| Лейкоциты (10 9 ) | 6.8-8.3 | 8.4-10.3 | 10.4-15.3 | >15.3 |
| Альбумин г/л | 34-44 | 25-33 | 21-24 |
*-Суммарная оценка свыше 11 баллов связана с высокой вероятностью летального исхода без трансплантации печени.
Неврологическая симптоматика болезни Вильсона частично обратима при терапии хелаторами и проведении трансплантации печени, что связано с необратимыми поражениями подкорковых ядер головного мозга токсическими концентрациями меди.
Госпитализация
ПОКАЗАНИЯ ДЛЯ ГОСПИТАЛИЗАЦИИ С УКАЗАНИЕМ ТИПА ГОСПИТАЛИЗАЦИИ 4
Показания для плановой госпитализации [1-4]:
· для уточнения диагноза;
· подбор схемы терапии;
· декомпенсация, резистентность к лечению, побочные эффекты терапии.
· контроль эффективности терапии (оценка метаболизма меди, степень фиброзирования печеночной паренхимы и нарушения психо-неврологических функций, с определением показаний для своевременного выполнения ортотопической трансплантации печени.
Информация
Источники и литература
Информация
ОРГАНИЗАЦИОННЫЕ АСПЕКТЫ ПРОТОКОЛА
Список разработчиков протокола с указание квалификационных данных:
1) Шарипова Майра Набимуратовна – доктор медицинских наук, детский гастроэнтеролог клинико-диагностического отделения РГКП «Научный центр педиатрии и детской хирургии».
2) Карсыбекова Ляйля Мауленовна – доктор медицинских наук, профессор, педиатр клинико-диагностического отделения РГКП «Научный центр педиатрии и детской хирургии».
3) Мухамбетова Гульнар Амерзаевна – кандидат медицинских наук, профессор кафедры нервных болезней №1, РГП на ПХВ «Казахский национальный медицинский университет имени С.Д. Асфендиярова».
4) Калиева Шолпан Сабатаевна – кандидат медицинских наук, доцент, заведующая кафедрой клинической фармакологии и доказательной медицины РГП на ПХВ «Карагандинский государственный медицинский университет», клинический фармаколог.
Указание на отсутствие конфликта интересов: нет.
Рецензенты:
Кульниязова Гульшат Матаевна – доктор медицинских наук, профессор кафедры Общей врачебной практики №1 с курсом коммуникативных навыков РГП на ПХВ «Западно-Казахстанский государственный медицинский университет им. М. Оспанова».
Указание условий пересмотра протокола: пересмотр протокола через 5 лет после его опубликования или при наличии новых методов с уровнем доказательности.



