Что такое пароксизмальная активность головного мозга у ребенка
а) Механизмы эпилептических припадков. Эпилептический разряд, являющийся основным электрофизиологическим проявлением эпилептического припадка, обычно состоит из ритмичных высокоамплитудных колебаний электрического потенциала. Как правило, его можно записать при ЭЭГ с кожи волосистой части головы, но в некоторых случаях он остается невыявленным даже при снятии ЭЭГ с поверхности головного мозга; это зависит от объема патологического очага, его геометрии и специфики активности (Bancaud и Talairach, 1975).
Это прямое указание на наличие патологической чрезмерной нейрональной активности, которую Hughlings Jackson постулировал как причину эпилепсии. Эпилептический разряд, записанный на коже головы или с поверхности коры, вызывается как патологической активностью отдельных нейронов, так и избыточной синхронизацией огромных клеточных популяций. Как схематически показано на рисунке ниже, пики на ЭЭГ отражают суммарные мембранные потенциалы, которые в свою очередь являются прямым следствием внутриклеточных процессов.
Самым ярким из них (при припадках фокальной природы) является выраженное увеличение площади мембранной деполяризации, вызываемое возбуждающими постсинаптическими потенциалами — пароксизмальный сдвиг мембранного потенциала. На межприступной ЭЭГ эти процессы соответствуют пикам у порогового уровня пораженных нейронов, за которыми следует длительная усиленная постполяризация. Во время припадка эта гиперполяризация исчезает, и повторные импульсы становятся неконтролируемыми. Это, в свою очередь, возбуждает соседние нейроны, которые начинают синхронно генерировать электрический разряд.
При генерализованных припадках могут действовать разные механизмы, но в случае судорожного припадка возникает длительная мембранная деполяризация. С другой стороны, при развитии неконвульсивных припадков, таких как абсансы, имеет место последовательность возбуждающих и тормозных потенциалов (Snead, 1995). Медленная волна на ЭЭГ, очевидно, отражает ГАМК-зависимое усиленное торможение, которое, во взаимодействии с повышенным возбуждением, способствует возникновению и поддержанию состояния эпилептической гиперсинхронии.
Низкопороговые кальциевые каналы в ретикулярных таламических нейронах — водителях ритма, вероятно, вызывают гиперсинхронные разряды типа спайк-волна при абсансной эпилепсии. Gloor и Fariello (1988) постулировали, что первичная генерализованная эпилепсия характеризуется диффузно гипервозбудимой корой, так что эпилептический разряд может запускаться при возбуждении таламической ретикулярной системы, которое вызывается входящим таламокортикальным импульсом; таким образом, для его возникновения необходимы и кора, и субкортикальные структуры.
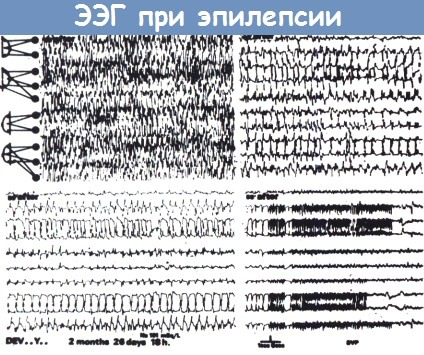 Генерализованный эпилептический припадок (у двухмесячного ребенка с гипернатриемией).
Генерализованный эпилептический припадок (у двухмесячного ребенка с гипернатриемией).
Тонический разряд (вверху слева), за которым следует клонический разряд (вверху справа, продолжение внизу слева).
Энцефалограмма, показанная внизу справа записана на медленной скорости и демонстрирует окончание припадка.
ЭЭГ-признаки этого припадка несколько атипичны, не одинаковы ритмы тонических разрядов правого и левого полушария.
Это часто встречается у детей; типичные генерализованные припадки у них наблюдаются редко.
Детальное описание механизмов, вызывающих эпилептический разряд, выходит за рамки настоящего руководства (для получения более подробной информации см. Jones, 2006; Najm et al., 2006b).
Повышенная возбудимость головного мозга может быть результатом генетически детерминированного химического дисбаланса между возбуждающими и тормозными нейромедиаторами, врожденных тонких изменений кортикальных нейрональных цепей или воздействия локального поражения. В этом отношении внимание исследователей привлекает возможное влияние нейронов, генерирующих патологические разряды на другие структуры головного мозга. При регулярно повторяющихся стимулах резко усиливается эффект фокальной субклинической стимуляции групп нейронов, что может привести к генерализованным припадкам (Moshe и Ludvig, 1988). Однако роль этого феномена «искры» в патогенезе эпилепсии у человека все еще не установлена, хотя этот процесс может быть фактором появления вторичных очагов (Morell, 1989).
Ослабленные механизмы торможения, как считается, играют важную роль в патогенезе эпилепсии; проводились широкие исследования основного кортикального тормозного нейромедиатора — гамма-аминомасляной кислоты (ГАМК). На основе гипотезы ГАМК разрабатываются основные терапевтические методики и антиэпилептические препараты.
В настоящее время исследования направлены на изучения таких возбуждающих нейромедиаторов, как глутамат и аспартат, избыток которых может вызывать локальное или генерализованное повышение кортикальной возбудимости (Naim et al., 2006b). Изменения рецепторов как возбуждающих (глутамат и аспартат), так и тормозных (ГАМК), вероятно, играют важную роль в регуляции кортикальной возбудимости, и различная скорость их «созревания» может объяснять различную предрасположенность к эпилепсии у разных возрастных групп (Moshe, 1987; Johnston, 1996; Holmes, 1997).
Вне зависимости от задействованных механизмов развитие головного мозга играет ключевую роль в предрасположенности к припадкам и их клинических проявлениях. В настоящее время активно изучаются морфологические, электрофизиологические и биохимические основы созревания головного мозга с точки зрения предрасположенности к припадкам (Jensen, 1999; Ben-Ari, 2006).

б) Причины пароксизмальных расстройств у детей. Симптоматические эпилептические припадки, как было сказано выше, могут вызываться большим количеством сопутствующих состояний, таких как высокая температура, гипогликемия, метаболический дисбаланс или острые заболевания. Почти всегда они проявляются как генерализованные судорожные припадки. Это может быть следствием таких состояний, как острое повреждение структур головного мозга вследствие травмы, метаболических расстройств или инфекций, которые могут поражать детей, не имеющих никакой предрасположенности к припадкам. Однако чаще все же имеется специфическая предрасположенность к припадкам, в основном, вероятно, генетического происхождения, и часто зависящая от возраста, и определяющая индивидуальную чувствительность к действию специфических стимулов; наиболее частым примером являются фебрильные судороги.
Тем не менее, с точки зрения этиологии нельзя противопоставлять эти две группы симптоматических припадков. Структурные нарушения гораздо чаще вызывают судороги у пациентов с наличием в семейном анамнезе случаев эпилепсии, среди населения в целом, то же самое относится и к другим причинам, например, острому менингиту.
Эпилепсия (как хроническое заболевание) также может вызываться только генетическими факторами или хроническим поражением головного мозга (врожденным или приобретенным) или различными комбинациями этих факторов (Berkovic et al., 1987).
Эпилепсии обычно подразделяют на идиопатические, симптоматические и криптогенные. При идиопатических эпилепсиях не удается обнаружить поражение или патологию головного мозга; симптоматические эпилепсии развиваются вследствие какой-либо патологии или приобретенного поражения; криптогенные эпилепсии — это те, при которых вероятно наличие органической причины, обнаружить которую, однако же, не удается. С развитием методов исследования последняя категория встречается все реже.
Генетические факторы играют важнейшую роль при эпилепсии, не связанной с неврологическими расстройствами (идиопатические эпилепсии), но также могут иметь значение при эпилепсии, связанной с выявленными повреждениями головного мозга (Ottman, 1989, 2005).
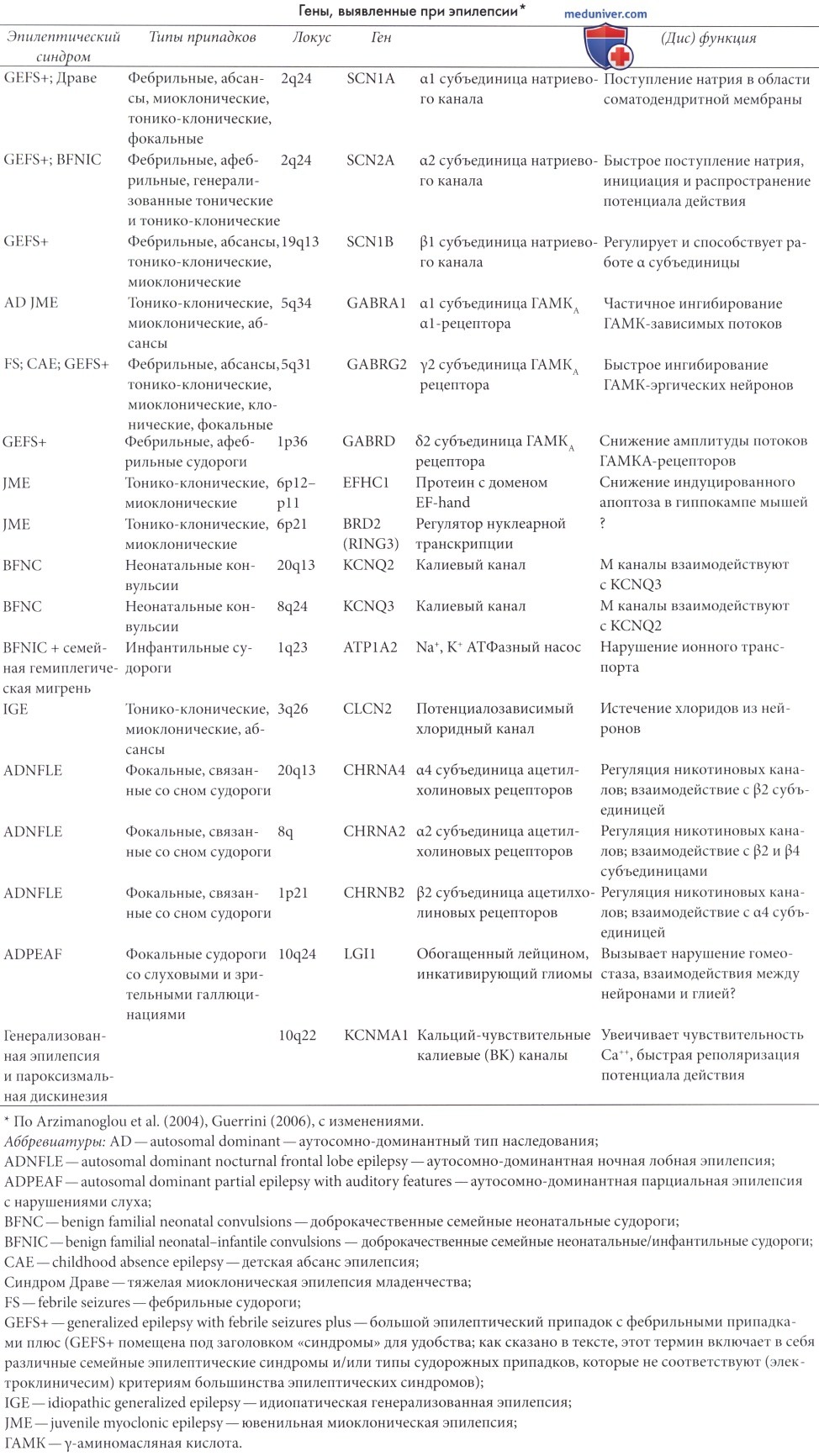
Их роль и тип наследования варьируют в зависимости от типа эпилепсии; далее в таблице ниже приведены данные по генетике каждого эпилептического синдрома. За последние 20 лет было установлено, что некоторые генетические синдромы имеют монофакториальное происхождение; в настоящее время бурно развивается молекулярная генетика эпилепсии у детей (подробнее см. Battaglia и Guerrini, 2005; Duron et al., 2005; Gardiner, 2005; Heron et al., 2007). Только небольшое количество случаев (1-2%) может быть отнесено к однолокусным аномалиям, таким, как туберозный склероз или к метаболическим нарушениям. Эпилептический синдром с моногенным доминантным типом наследования, описанный ниже, включает в себя доброкачественные неонатальные конвульсии (Leppert et al., 1989), доброкачественные инфантильные судороги (Vigevano et al., 1992; Fxhenne et al., 1994), семейный лобный (Scheffer et al., 1995a), медиальный и латеральный (Berkovic et al., 1996; Andermann и Kobayashi, 2005), височно-долевой эпилептические синдромы.

Действие генетических факторов не ограничивается этими относительно редкими случаями. Было выявлено несколько генов, участвующих в развитии немоногенных форм эпилепсии. Гораздо чаще встречается комплексное наследование (Turnbull et al., 2005), хотя для эпилепсий с 3 Гц спайк-волновой активностью и, возможно, некоторых форм фотосенситивной эпилепсии предполагается доминантный механизм наследования (Doose и Waltz, 1993). Doose и Baier (1987) предположили, что определенные ЭЭГ-признаки (например, фоточувствительность или спонтанные пароксизмы спайк-волн) могут наследоваться независимо, и что комбинация таких ЭЭГ-признаков (возможно, и других факторов) может указывать на наследственную предрасположенность к различным формам эпилепсии.
Считалось, что некоторые синдромы, для которых был определен доминантный ген, например, ювенильная миоклоническая эпилепсия, наследуются по доминантному типу, однако в настоящее время выявлено, что эти состояния наследуются по комплексному (многофакторному) механизму (Greenberg et al., 1988), и в наследовании участвуют несколько других генов.
В настоящее время считается, что многие гены наследования эпилепсии—это гены предрасположенности, чье сочетанное действие и дополнительные факторы среды увеличивают вероятность возникновения клинической эпилепсии; это объясняет тот факт, что сиблинги/родители больных могут не страдать эпилепсией или иметь другие формы эпилептического синдрома.
Повреждение головного мозга является основным этиологическим фактором парциальной или генерализованной эпилепсии, сочетающейся с клиническими проявлениями дефектов развития нервной системы. Причины включают в себя опухоли головного мозга, травмы, нарушения развития коры, туберозный склероз, поражения, связанные с синдромом Штурге-Вебера, склероз гиппокампа и сосудистые аномалии. Основные этиологические факторы поражений головного мозга, сочетающихся с эпилепсией, перечислены в таблице ниже.
Определение симптоматическая или «связанная с поражением головного мозга» используется в случаях, когда поражение вызывает клинические нарушения или выраженные изменения, выявляемые при визуализации и/или когнитивные нарушения. Микроскопические изменения, при жизни обычно не выявляемые, также играют важную, хотя и не определенную до сих пор роль в патогенезе эпилепсии (так называемые криптогенные эпилепсии). С помощью современных методов нейровизуализации было выявлено, что многие случаи криптогенной эпилепсии на самом деле вызваны внутричерепным патологическим процессом (подробнее см. Arzimanoglou et al., 2004). Патология развития коры наиболее вероятная причина так называемых криптогенных фокальных эпилепсий.
Однако механизм развития эпилепсии при наличии повреждения, и такие феномены, как период латентности между поражением и началом припадков, все еще не совсем понятны.
Эпилепсия также является частым проявлением таких хромосомных расстройств, как синдром Ангельмана, синдром Дауна, трисомия 12p, кольцевые хромосомы 14 и 20, синдром ломкой Х-хромосомы, синдром del 1p36 (синдром делеции короткого плеча 1 хромосомы — прим, перге.) (Battaglia и Guerrini, 2005), и также может быть связана с врожденными нарушениями метаболизма (Wolf et al., 2005).
Редактор: Искандер Милевски. Дата публикации: 3.1.2019
Что такое пароксизмальная активность головного мозга у ребенка
Клиническая картина церебральных пароксизмов как эпилептического, так и неэпилептического генеза отличается многообразием проявлений, что приводит порой к диагностическим ошибкам и назначению неадекватной терапии. Следует помнить, что гипердиагностика эпилепсии в перспективе может причинять не меньше вреда пациенту, чем пропущенные случаи. Большинства диагностических ошибок можно избежать благодаря тщательному и подробному сбору анамнестических сведений, грамотному подходу к наблюдению пациента и адекватной оценке инструментальных методов обследования. Описываемый нами случай демонстрирует полиморфизм пароксизмальных состояний в детском возрасте.
Клинический случай
Пациентка В., поступила в неврологическое отделение 3-й ГДКБ Минска в возрасте 1 год 6 месяцев с жалобами на эпизодическое «закатывание» глаз в сторону в течение нескольких секунд, при этом в момент реализации данного пароксизма у ребенка отсутствовала реакция на обращение и звуковые раздражители. Подобные состояния отмечались мамой девочки в течение недели до момента поступления в стационар. Заболевание началось с 2-х эпизодов в течение суток, затем количество пароксизмов увеличилось до 10–15 раз. Предшествующее заболеванию травматическое повреждение центральной нервной системы мамой отрицалось; ранее у ребенка в анамнезе также не отмечалось черепно-мозговых травм, нарушений сознания либо пароксизмальных состояний эпилептического и неэпилептического генеза.
Из анамнеза: ребенок от 2-й беременности, первых срочных родов. Родоразрешение было осуществлено путем вакуум-экстракции в сроке 41 неделя гестации. Беременность протекала на фоне угрозы прерывания по причине ОРВИ в 1-м и 2-м триместрах. Вес при рождении составлял 3 810 г, длина 51 см. Вскармливание искусственное. Развитие ребенка до госпитализации соответствовало возрастной норме, прививки выполнены. За предшествующий период ребенок переносил только простудные заболевания. Хроническую соматическую патологию и оперативные вмешательства мама отрицала. Наследственность и трансфузионный анамнез не отягощены, однако у девочки наличествовала аллергологическая патология (пищевая аллергия на белок коровьего молока).
При поступлении соматический статус без особенностей. Физическое развитие среднее дисгармоничное по весу. Неврологический статус: девочка в сознании, эмоционально лабильна, в поведении адекватна, доступна осмотру и соответствующему возрасту контакту. Сон, со слов матери, спокойный. Со стороны черепных нервов отмечалась легкая асимметрия носогубных складок. Верхние конечности: объем движений и мышечный тонус физиологический. Мышечная сила соответствует возрасту. Трофика мышц без особенностей. Гиперкинезов нет. СПР D=S, оживлены, рефлексогенные зоны не расширены. Брюшные рефлексы D=S, средней живости. Нижние конечности: объем движений физиологический. Мышечная сила соответствует возрасту. Трофика мышц без особенностей. Мышечный тонус ближе к физиологическому, слегка повышен. Гиперкинезов нет. Рефлексы коленные D=S, ахилловы D=S. Патологических стопных знаков нет. Координаторная сфера без особенностей. Ходит самостоятельно с 1 года 1 месяца, несколько ротирует внутрь правую стопу, опора с тенденцией на передний отдел стоп (эпизодическая доброкачественная дистоническая реакция опоры на стопу). Менингеальных симптомов нет. Тазовые органы без патологии.
На вторые сутки пребывания в отделении количество эпизодов отведения глаз в сторону составило порядка 10. Длительность эпизодов колебалась от 5–10 секунд до нескольких минут. Эпизодам отведения глаз сопутствовало расстройство координации легкой степени выраженности, которое исчезало через 5–10 минут после окончания окуломоторного пароксизма.
Выставлен предварительный диагноз: пароксизмальные состояния с наличием эпизодов тонического отведения глаз в сторону неуточненного генеза.
До проведения обследования с целью снижения возбудимости центральной нервной системы ребенку назначен громецин в дозе 0,1 г по 1 таблетке 2 раза в день.
Проведенное электроэнцефалографическое (ЭЭГ) обследование выявило легкие диффузные изменения с признаками нейрофизиологической незрелости. Пароксизмальной активности не зарегистрировано (см. рис. 1). Общий и биохимический анализы крови без патологии.
Рисунок 1. Первичное ЭЭГ-обследование. Легкие диффузные изменения с признаками нейрофизиологической незрелости. При проведении нейровизуализационного исследования (МРТ 2 Тл) выявлены признаки некоторого усиления МР-сигнала в парагиппокампальной области (см. рис. 2а) и незначительного расширения переднего субарахноидального пространства (см. рис. 2б), что может свидетельствовать в пользу перенесенной гипоксически-ишемической перинатальной энцефалопат На третьи сутки количество эпизодов отведения глаз сократилось до 6, при этом их продолжительность не превышала нескольких секунд.
Рисунок 2а. МРТ-исследование. Признаки усиления МР-сигнала в парагиппокампальной области.
Рисунок 2б. МРТ-исследование. Признаки незначительного расширения переднего субарахноидального пространства. На четвертые сутки количество пароксизмов продолжило уменьшаться и составило всего 2 эпизода длительностью 1–2 секунды. При этом ранее сопутствовавших данным состояниям нарушений координации не отмечалось.
На пятые сутки эпизоды отведения глаз полностью прекратились и в последующие дни пребывания в стационаре не повторялись.
Повторные ЭЭГ-обследования, проведенные на третьи и шестые (см. рис. 3а, 3б) сутки от момента госпитализации, региональных и патологических форм биоэлектрической активности не выявили.
Исследование крови на вирусы, бактерии и простейшие не позволило выявить значимого инфекционного фактора в развитии текущего заболевания.
Рисунок 3а. Повторное ЭЭГ-обследование. Легкие диффузные изменения биоэлектрической активности головного мозга. Региональных и патологических форм активности не выявлено.
Рисунок 3б. Повторное ЭЭГ-обследование. Легкие диффузные изменения биоэлектрической активности головного мозга. Региональных и патологических форм активности не выявлено.
Окончательный диагноз: пароксизмальные состояния по типу доброкачественных эпизодов тонического отведения глаз в сторону неэпилептического генеза.
Противоэпилептическая терапия не назначалась. Ребенок выписан домой в удовлетворительном состоянии.
При контрольном осмотре через 2 месяца после выписки из стационара было установлено, что пароксизмальные состояния не повторялись, неврологический статус сохранялся прежним. Девочка продвинулась в психоречевом развитии: увеличился активный и пассивный словарный запас, стала говорить предложениями из двух слов. ЭЭГ-обследование существенных изменений не выявило.
Комментарий
Доброкачественные тонические отведения глаз в сторону у детей — это гетерогенная группа состояний с пролонгированными эпизодами постоянных или преходящих девиаций глазных яблок в противоположное желаемому направление с быстрым возвратом в исходное положение.
Этиология описываемого пароксизмального состояния неоднородна, а патогенез до настоящего времени остается до конца не установленным. По результатам исследований некоторых авторов, заболевание может иметь аутосомно-рецессивную и аутосомно-доминантную природу наследования. У таких пациентов могут выявляться мутации в гене кальциевого канала CACNA1A. Нельзя исключить вариант функционального генеза доброкачественного тонического отведения глаз в сторону вследствие незрелости мозговых структур и нарушения межнейронной коммуникации.
Наиболее часто исследователями рассматривается гипотеза возраст-зависимой дисфункции супрануклеарных путей. Этиологическими факторами симптоматических форм заболевания могут служить повреждения мезенцефальной области — мальформация вены Галена, пинеалома, гидроцефалия, опухоль гипофиза. Возможна также ятрогенная природа данного пароксизмального состояния. Ряд авторов указывает на более частое его развитие у тех детей, матери которых во время беременности принимали препараты вальпроевой кислоты.
Клинические проявления доброкачественного тонического отведения глаз в сторону отличаются некоторым разнообразием. Дебют заболевания, как правило, возникает в возрасте 4–10 месяцев. В наиболее крупном исследовании данной патологии возраст дебюта варьировал от 1 недели до 26 месяцев жизни (в среднем 5,5 месяца). Частота приступов вариабельна в течение суток от единичных до ежечасных. Характерно учащение и усиление выраженности пароксизмов при инфекционных заболеваниях, сопровождающихся лихорадкой, а также при физическом и психоэмоциональном переутомлении. Как правило, в течение всего периода заболевания модификации вида пароксизмов не происходит. Считается, что продолжительность заболевания и срок достижения ремиссии (спонтанного исчезновения отведения глаз) в среднем могут варьировать от нескольких дней до 5 лет.
При доброкачественных тонических отведениях глаз в сторону и вверх у ряда детей отмечаются эпизоды дополнительного наклона подбородка вниз, однако ряд авторов полагает, что это компенсаторный механизм с целью коррекции неправильной позиции глазных яблок. При существующем стереотипе отведения глаз в определенном направлении противоположное движение обычно не изменено. Тем не менее, у части пациентов во время пароксизма может иметь место однонаправленный нистагм с быстрым компонентом в обратную сторону.
Кроме того, некоторыми исследователями описаны гипометрические саккады и расходящееся косоглазие, которые могут сохраняться в течение некоторого времени (в отдельных случаях — длительно) после прекращения тонических отведений глаз.
Описываемое неэпилептическое пароксизмальное состояние периодически сопровождается моторной неловкостью и атаксией различной степени выраженности, продолжительностью от нескольких часов до нескольких дней. Координаторная дисфункция легкой степени выраженности также может иметь тенденцию к сохранению после разрешения основного заболевания.
По данным различных авторов, около 50 % детей с доброкачественным тоническим отведением глаз в сторону имеют нормальное психическое развитие, приблизительно 40 % — легкий интеллектуальный дефицит, 10 % — нарушения интеллекта от умеренной до выраженной степени. В подавляющем большинстве случаев нормальный психоневрологический статус у пациентов с описываемым вариантом пароксизмов коррелирует со спонтанной ремиссией в течение 2 лет от начала заболевания. Если же имеет место сочетание данного состояния и неврологического и/или когнитивно-мнестического дефицита, то необходимо дополнительное обследование ребенка на предмет симптоматической природы этиологии патологического процесса.
Инструментально-диагностические и лабораторные исследования чаще всего демонстрируют нормальные показатели нейровизуализации, электроэнцефалографии и метаболических параметров крови и ликвора у детей с идиопатическими (генетическими) формами заболевания. Золотой стандарт нейровизуализационного обследования — МРТ головного мозга, как правило, не выявляет патологических изменений.
Дифференциальный диагноз доброкачественных тонических отведений глаз в сторону проводят с эпилептическими пароксизмальными состояниями, в частности с окуломоторными приступами и атипичными абсансами.
Специфическое лечение данного состояния не разработано, попытки терапевтического воздействия чаще всего оказываются неэффективными. По данным наиболее крупного исследования, терапия антиконвульсантами, ацетазоламидом и адренокортикотропным гормоном не оказала влияния на течение заболевания. Тем не менее ряд авторов указывает на достижение положительного эффекта в отношении частоты и выраженности пароксизмов при применении дигидроксифенилаланина (леводопы).
Пароксизмальные дискинезии: дифференциальный диагноз с эпилепсиями
Пароксизмальные дискинезии — это неврологические состояния с разнообразной клинической картиной, характеризующиеся внезапными атаками патологической непроизвольной двигательной активности (т. е. сопровождающиеся приступами гиперкинезов) в мышцах конечнос
Пароксизмальные дискинезии — это неврологические состояния с разнообразной клинической картиной, характеризующиеся внезапными атаками патологической непроизвольной двигательной активности (т. е. сопровождающиеся приступами гиперкинезов) в мышцах конечностей, туловища, лица, шеи [3]. Приступы гиперкинезов мы называем здесь «атаками гиперкинезов», чтобы не смешивать с термином «эпилептические приступы».
Уместность термина «пароксизмальные» обусловлена тем, что гиперкинезы внезапно появляются и также внезапно исчезают, принимая форму атак. Между атаками гиперкинезов двигательная сфера и поведение человека в целом остаются совершенно нормальными. Атаки гиперкинезов могут проявляться непроизвольными быстрыми нерегулярными подергиваниями (хореей); медленными змееподобными или червеобразными движениями, плавно перетекающими от одних мышц к другим (атетозом); повышением мышечного тонуса с повторными выкручивающими движениями и патологическими позами (дистонией); неконтролируемыми круговыми движениями в одной или нескольких конечностях (баллизмом) или любым сочетанием перечисленных гиперкинезов.
Пароксизмальные дискинезии делят на пароксизмальную кинезигенную дискинезию (ПКД) и пароксизмальную некинезигенную дискинезию (ПНКД). Атаки гиперкинезов при ПКД развиваются в результате провоцирующих факторов (триггеров), которые действуют неожиданно и внезапно. Напротив, при ПНКД атаки происходят спонтанно в состоянии покоя или прерывают повседневную двигательную активность; их выраженность усиливают алкоголь, кофе, стресс, бурные эмоции и т. д. [1]. Отдельно выделяют два вида пароксизмальных дискинезий: провоцирующиеся длительной или чрезмерной физической нагрузкой (ПКФН) и провоцирующиеся сном (гипногенная дискинезия — ПГД).
Пароксизмальные дискинезии характеризуются в зависимости от продолжительности атак (атаки бывают непродолжительными — менее 5 мин или длительными — более 5 мин), могут носить семейный характер, развиваться вследствие неясных причин (спорадические формы) или вторично, на фоне какого-либо заболевания (симптоматические формы).
ПКД раньше назывались пароксизмальным хореоатетозом, но современный термин более корректен, поскольку при ПКД развивается не только хореоатетоз, но и другие виды гиперкинезов. У многих пациентов с ПКД заболевание является идиопатическим (т. е. семейным или спорадическим), симптомы манифестируют до 20 лет, особенно часто — до 10 лет. В целом возраст дебюта варьирует от 6 мес до 40 лет. По литературным данным, мальчики болеют чаще девочек, но точная распространенность неизвестна из-за того, что заболевание встречается крайне редко.
Транзиторные атаки гиперкинезов при ПКД на ранних стадиях затрагивают мышцы рук и ног, но по мере течения заболевания, гиперкинезы охватывают также мышцы лица, шеи и туловища. Атаки могут быть односторонними или двусторонними, однако характерной является их несимметричность даже при билатеральном характере. Непроизвольное сокращение лицевых или оромандибулярных мышц проявляется гримасничаньем, нечеткостью речи (дизартрия) и даже мутизмом. Атаки никогда не сопровождаются изменением сознания. Гиперкинез, возникающий в ногах или туловище, приводит к внезапному падению и зачастую многочисленным травмам. Кроме внезапных двигательных атак, появляются автоматизмы — зевание, одышка, эхолалии, эхопраксии. Атаки учащаются под действием внешних факторов.
У большинства пациентов с ПКД атаки отмечаются днем. Их частота различна — от 1 в месяц или несколько месяцев до 100 ежедневно. В начале каждой атаки у некоторых пациентов возникают ощущения-предвестники (покалывание, жжение и другие парестезии; головокружение; мышечный спазм). После этого, как правило, развивается гиперкинез на том же участке тела.
Частота атак при ПКД с возрастом уменьшается. У большинства пациентов с ПКД атаки короткие — от нескольких секунд до 5 мин. Реже атаки гиперкинезов длятся несколько часов. ПКД с длительными и редкими атаками дебютируют в более позднем возрасте. Продолжительность атак может меняться. Приведем пример.
Девочка, П. К., 6 лет. Диагноз: аутосомно-рецессивная роландическая эпилепсия. Пароксизмальная дискинезия. Жалобы: пароксизмы гиперкинезов в левых конечностях и левой половине лица. Анамнез жизни: наследственность отягощена: по линии матери (у бабушки три двойни, недоношенность, выкидыши). Ребенок от первой беременности, протекавшей на фоне угрозы прерывания на ранних сроках, в 4 мес — кровянистые выделения (стационарное лечение), на фоне повторных острых респираторных вирусных инфекций (ОРВИ); первые срочные стремительные роды. Закричала сразу. Выписана на 7-е сутки. На естественном вскармливании до 1,5 мес. Вес набирала хорошо. Психомоторное развитие: голову держит — к 3 мес, сидит — к 7 мес, ходит — к 11 мес. Говорит соответственно возрасту.
Анамнез заболевания: в возрасте 1 года при каждом пробуждении появились подергивания левой ногой (несинхронные, иногда вращательные движения), через 2–3 мес подергивания стали возникать одновременно в ноге и руке, спустя несколько месяцев присоединились подергивания в лице с той же стороны. В 1,5 года поставлен диагноз — эписиндром, левосторонние джексоновские судороги. Назначены глюферал, затем конвульсофин, депакин, бензонал в виде моно- и политерапии (до трех препаратов одновременно) — без эффекта. На фоне лечения частота атак сохранялась от 1 до 15 в сутки, они возникали не только при пробуждении, но и во время бодрствования (на провокацию эмоциональной и физической нагрузкой); во время сна в фазe дремоты, перед пробуждением. Максимальная длительность до 30 с. Сознания не теряла. Атаки предчувствует.
В неврологическом статусе — очаговой симптоматики не выявлено.
Видеоэлектроэнцефалография (видео-ЭЭГ): фоновая ЭЭГ при закрытых глазах в затылочных отделах представлена гиперсинхронной альфа-активностью, амплитудой 150–200 мкВ, частотой 8 Гц. При проведении фотостимуляции частотой 18 Гц отмечаются генерализованные вспышки комплексов «пик-волна», не сопровождающиеся клиническими паттернами эпилептического припадка. Длительность вспышки — 1–3 с. На ЭЭГ сна зарегистрированы 1–3-я стадии сна. В 1–2-й стадиях регистрируются двухфазные острые волны, локализующиеся в центральной области с амплитудным преобладанием справа. Во сне также зарегистрированы генерализованные вспышки эпилептического характера, не сопровождающиеся клиническими проявлениями. При пробуждении и в состоянии бодрствования зафиксированы два эпизода движений в левой руке в виде хореоатетоза. Моторный феномен при пробуждении не сопровождался изменениями биоэлектрической активности (БЭА). Второй феномен совпал со вспышкой генерализованной эпилептической активности. Заключение: при видео-ЭЭГ-мониторинге выявлено два типа патологической активности: 1) локальные изменения в центральной области, которые могут быть характерны для роландической эпилепсии; 2) генерализованная эпилептическая активность. Двигательные феномены носили характер гиперкинезов (хореоатетоз) и, вероятно, не являются эпилептическими приступами.
Результаты обследования: биохимический анализ крови, электрокардиограмма (ЭКГ), ультразвуковое исследование (УЗИ) внутренних органов: без особенностей. Магнитно-резонансная томография (МРТ) головного мозга — без патологии. Офтальмолог: без изменений. Психолог: психологическое развитие соответствует возрастной норме. Обследована в лаборатории наследственных болезней обмена веществ медико-генетического центра РАМН: исключены GM1-ганглиозидозы, нейрональный цероидный липофусциноз 1 и 2, митохондриальные заболевания, болезнь Вильсона–Коновалова.
На фоне отмены противосудорожной терапии (карбамазепин, барбитураты) изменения частоты приступов не произошло. Введен наком. Приступы хореоатетоза в левых конечностях сохранялись с частотой 5–11 раз в сутки, в дневное и ночное время, периодически были связаны с движениями и эмоциональной нагрузкой (табл. 1). После выписки частота приступов снизилась. Спустя 6 мес проведена коррекция терапии (увеличение дозы накома, фризиума), приступы прекратились. В течение 1 мес приступов не было, затем они вновь возобновились, в основном в утренние часы, в связи с пробуждением, реже во время бодрствования.
 |
| Таблица 1. Дневник эпизодов в течение одного дня |
ЭЭГ на фоне терапии в катамнезе: отмечается положительная динамика в виде снижения представленности диффузных эпилептиформных разрядов, снижения амплитуды региональных центрально-вертексных разрядов, а также их урежения во время бодрствования.
ПНКД. Прежнее название — «пароксизмальный дистонический хореоатетоз». ПНКД чаще манифестирует до 20 лет, но возраст дебюта крайне вариабелен. Чаще болеют мальчики. Атаки характеризуются наличием любых гиперкинезов (хореи, атетоза, дистонии, баллизма). Необходимо подчеркнуть, что они никогда не сопровождаются нарушением сознания. Атаки бывают столь тяжелыми, что вызывают внезапное падение или нарушение походки и других видов двигательной активности, обусловливая тяжесть заболевания. Атаки при ПНКД развиваются внезапно, без каких-либо специфических триггеров, что отличает их от ПКД. Тяжесть атак может усугубляться при испуге, волнении, возбуждении, радости, охлаждении или перегревании, употреблении алкоголя, кофе, чая, шоколада и т. д. Частота атак ПНКД значительно реже, чем при ПКД, 2–3 раза в месяц; однако иногда они развиваются более 100 раз в день. Атакам предшествуют предвестники в виде необычных ощущений (покалывание, разливающееся тепло и др.), мышечное напряжение. Атаки при ПНКД длятся от 5 мин до нескольких дней. По длительности их подразделяют на короткие (менее 5 мин) и длительные (более 5 мин). С возрастом длительность атак постепенно уменьшается.
ПКФН — редкая форма пароксизмальных дискинезий, при которых атаки провоцируются длительной или чрезмерной физической нагрузкой (прежнее название — «промежуточная пароксизмальная дискинезия»).
У пациентов с идиопатической (семейной или спорадической) формой заболевание дебютирует в детском возрасте. Лишь при вторичных (симптоматических) формах возраст дебюта увеличивается до 30 лет. Чаще болеют девочки.
ПКФН характеризуется прежде всего внезапными транзиторными дистоническими атаками, проявляющимися непроизвольным повторным сокращением мышц с формированием патологических поз (часто болезненных). У некоторых пациентов атаки дистонии проявляются хореоатетозом. Атаки возникают на фоне сильной или длительной физической нагрузки (бег, прогулки на большие расстояния), иногда ПКФН провоцируются пассивными движениями в пораженных конечностях, усиливаются под действием внешних факторов. Атаки в основном возникают в ногах, но иногда, особенно при длительных эпизодах, вовлекаются мышцы лица, шеи, туловища. Как правило, поражаются симметричные части тела (билатеральные атаки), но бывают и асимметричные. Ощущений-предвестников при ПКФН не отмечено. Частота атак составляет 1–5 в месяц. Описаны случаи, когда атаки происходят 1–2 раза в день. Продолжительность атак — от 5 до 30 мин, реже менее 2 мин. Длительность атак уменьшается с возрастом, в целом они продолжаются от нескольких секунд (у взрослых) до нескольких дней (у детей). Приведем второй пример.
Девочка, К. Д., 9 лет. Диагноз: пароксизмальная кинезигенная дискинезия. Симптоматическая эпилепсия. Детский церебральный паралич (ДЦП), спастико-гиперкинетическая форма. Жалобы: отставание в психомоторном развитии, периодические приступы червеобразных насильственных движений, длящиеся от нескольких часов до суток. Анамнез жизни: ребенок от молодых здоровых родителей, первых преждевременных родов на 36-й неделе. При рождении вес — 3,0 кг, рост — 50 см. Развитие с задержкой психомоторных навыков. В 8 мес поставлен диагноз ДЦП. Систематически получает восстановительную терапию. Наследственность не отягощена.
Анамнез заболевания: в возрасте 7 лет на фоне ОРВИ развилась первая атака гиперкинезов в виде хореоатетоза, продолжавшаяся 3 сут. Из-за тяжести состояния (дыхательная аритмия, непрекращающиеся гиперкинезы) ребенок находился в отделении реанимации. На фоне терапии (тизерцин, финлепсин, депакин, циклодол) гиперкинезы начали ослабевать к концу 3-х суток и затем прекратились. Двигательная активность полностью восстановилась. Через 2–3 нед — повторная атака хореоатетоза, продолжительностью около 10 мин, менее тяжелая, без утраты сознания. Затем атаки гиперкинезов, продолжительностью несколько часов, повторялись 1 раз в месяц.
Неврологический статус: легкое расходящееся косоглазие, слабость конвергенции слева. Девиация языка вправо. Походка спастико-паретическая с пропульсией. Гемиатрофия конечностей справа. Мышечный тонус повышен по спастическому типу, с элементами пластики. Сухожильные и периостальные рефлексы повышены с расширением зон, выраженнее справа. Двусторонний клонус стоп. Патологические стопные рефлексы с обеих сторон. В позе Ромберга по тяжести состояния не исследована. Координаторные пробы выполняет с интенцией и дисметрией слева. Расстройств чувствительности не выявлено. Дистонические атаки в ответ на эмоциональную или физическую провокацию.
Результаты обследования. ЭКГ: без патологии. Биохимический анализ крови: без патологии. Офтальмолог: передний отрезок, преломляющие среды, глазное дно — без изменений. Нейропсихолог: дефицит кинестетической организации движений и речи, а также модально-специфические нарушения слухоречевой памяти и слабость предметного зрительного гнозиса. ЭЭГ-мониторинг дневного сна: в левой центрально-теменной области регистрируются единичные монофазные острые волны, частотой 1–2 в секунду, с последующим периодом учащения острых волн до 5 в секунду и появлением в их ряду отдельных комплексов «пик-волна». С учетом данных обследования атаки были расценены как приступы симптоматической эпилепсии; назначен депакин (600 мг/сут). На фоне депакина в течении 6 мес отмечалось полное отсутствие атак. На фоне острого ринофарингита появились гиперкинезы, усиливающиеся по интенсивности, частоте и амплитуде в течение 2 сут. Введение диазепама, затем аминазина не позволяли контролировать атаки гиперкинезов, которые повторялись каждые 5–10 мин и носили «статусный» характер, в связи с чем ребенок переведен в отделение реанимации. В отделении реанимации в связи с некупирующимися дистоническими атаками (баллизм, хореоатетоз) было принято решение о введение тиопентала, миорелаксантов, искусственной вентиляции легких (ИВЛ) по жизненным показаниям. Течение ОРВИ осложнилось явлениями верхнедолевой пневмонии. На фоне введения тиапридала внутривенно в дозе 300 мг/сут, в течение 5 дней дистонические атаки сохранялись. На 6-е сутки внутривенно введен аминазин 1,0 мл. Гиперкинезы продолжались. Внутривенное введение депакина, диазепама, натрия оксибутирата прекращали гиперкинезы на непродолжительное время. На 8– 10-е сутки проведена пульс-терапия метилпреднизолоном в дозе 400 мг/сут без эффекта. На 11-е сутки применили наком 1/8 таблетки (утром). Лечение в течениие 4 дней без эффекта. На 15-е сутки перорально назначены клоназепам — до 1,5 мг/сут, топамакс — 50 мг/сут, финлепсин — 300 мг/сут — без эффекта. На фоне временного прекращения инфузий тиопентала в течение 2 ч хореоатетоз сохранялся. Ребенок находился в сознании, реагировал на осмотр. Прекратить инфузии тиопентала не удавалось. На 15-е сутки к тиапридалу в дозе 600 мг/сут добавлен обзидан в дозе 0,5 мг/сут (на физрастворе 20,0 — через инфузомат), на следующий день проведена коррекция дозы обзидана — 0,7 мг/сут на физрастворе 20,0 (1 мл/ч) с отчетливым положительным эффектом в виде урежения гиперкинезов, появления «светлых промежутков». На 17-е сутки сохранялись лишь постоянные миоклонии конечностей и плечевого пояса. Доза обзидана доведена до 1 мг/сут. ЭЭГ-мониторинг в отделении реанимации: диффузные функционально-органические нарушения БЭА головного мозга. Данных, свидетельствующих о наличии региональной, генерализованной, диффузной эпилептиформной активности, нет. Зарегистрированные в ходе исследования многочисленные миоклонии, с учетом клинико-электроэнцефалографических коррелятов, расценивались как миоклонус неэпилептического генеза. В результате МРТ головного мозга выявлены: базально-височная атрофия слева, вторичное расширение базальных субарахноидальных пространств в данной области до степени кисты; диффузная корково-подкорковая субатрофия, реализованная диффузным расширением субарахноидальных пространств конвекситальных отделов коры и реактивным асимметричным, преимущественно правосторонним расширением боковых желудочков. Обследована генетиком: исключены GM1- и GM2-ганглиозидозы, NARP (нейропатия, атаксия, пигментный ретинит), органические ацидурии и аминоацидопатии, митохондриальные болезни, болезнь Вильсона–Коновалова, болезнь Галлевордена–Шпатца. На фоне терапии (тиопридал, обзидан, депакин) был прекращен тиопенталовый наркоз, гиперкинезы редуцировались. В целом длительность атаки составила 17 суток. В стабильном состоянии пациентка переведена из реанимации в отделение психоневрологии, где получала депакин — 600 мг/сут, анаприлин — 10 мг/сут, ноотропил — 600 мг 2 раза в сутки, тиапридал — 100 мг/сут. На фоне однократного снижения дозы тиапридала до 3,3 мг в утренний прием отмечалось усиление гиперкинезов. К моменту выписки из стационара неврологический статус ребенка не отличался от состояния при поступлении. Сохранялись небольшие дистонические атаки на провокацию. Наблюдение в катамнезе составило 3 года.
ПГД — редкий вариант болезни, характеризующийся транзиторными атаками непроизвольных движений во время NREM-фазы сна (фаза медленного сна). Изредка атакам ПГД предшествуют специфические ощущения-предвестники. Атаки часто связаны с периодами пробуждения (arousal); происходят во сне, при этом глаза больного открыты, а в конечностях и туловище возникают гиперкинезы. Иногда атаки сопровождаются вокализациями, нарушением дыхательного ритма, тахикардией. Затем продолжается нормальный сон, сами атаки полностью амнезируются. Тяжесть атак усугубляется под воздействием внешних факторов [4].
Идиопатические варианты дебютируют в детстве, причем семейные формы раньше, чем спорадические. Возраст дебюта варьирует от 2 до 23 лет — при семейных случаях и от 3 до 47 лет — при спорадических. Частота атак обычно составляет 4–5 раз в год, но иногда она возрастает до 4–5 раз за ночь. Атаки обычно короткие — от 20–50 с до 2 мин.
У некоторых пациентов наряду с ночными атаками отмечаются атаки в период бодрствования, кинезигенные или некинезигенные. Кроме того, при семейных вариантах у разных членов семьи могут отмечаться разные формы пароксизмальных дискинезий. Приведем еще один пример.
Ребенок, И. О., 6 лет. Диагноз: ДЦП, левосторонний гемипарез. Роландическая эпилепсия. Пароксизмальная дискинезия. Жалобы при поступлении на эпизоды «потягиваний» во время сна при перемене положения тела (тоническое напряжение в мышцах шеи и спины с последующим разгибанием туловища и запрокидыванием головы). Пароксизмы сопровождаются прекращением дыхания, вегетативными симптомами в виде похолодания кожи, оперкулярными автоматизмами (причмокивания). В утренние часы после ночных пароксизмов вялый, неловкий, спотыкается. Частота пароксизмов — 1 раз в месяц.
Из анамнеза: родители здоровы, наследственность не отягощена. Ребенок от 4-й беременности, характеризующейся патологическим течением (на фоне хронического гайморита, отита, угрозы прерывания с отслойкой плаценты во II триместре, гипотиреоза, резус конфликта), со стационарным лечением (гормональные препараты); вторых преждевременных родов на
34-й неделе путем экстренного кесарева сечения. Вес при рождении 2,7 кг, рост 52 см. Ребенок родился в тяжелой асфиксии, 7 дней находился на ИВЛ, перенес пневмонию. В связи с гемолитической болезнью новорожденных однократно проводилось заменное переливание крови. По данным нейросонографии, в период новорожденности отмечалось кровоизлияние в боковые желудочки мозга с исходом в перивентрикулярную лейкомаляцию. С рождения наблюдается у невролога. С 2 лет отмечаются ночные приступы в виде болей в животе, позывов на дефекацию, сопровождающихся перекосом лица и оперкулярными движениями в течение получаса, с последующим постприступным сном. С 3 лет приступы видоизменились (простые фокальные моторные в виде клонических судорог в левой верхней конечности продолжительностью 15 мин с последующим сном). Был назначен финлепсин ретард — 300 мг/сут, после чего появились ночные «потягивания» при перемене положения тела во сне. На фоне введения и увеличения дозы депакина хроно до 1000 мг/сут «потягивания» стали реже. В связи с развитием тромбоцитопении доза депакина хроно была снижена до 750 мг/сут и увеличена доза финлепсина до 400 мг/сут. В 4 года на фоне герпетического стоматита развились генерализованные тонико-клонические приступы с потерей сознания, частота приступов — 5 раз за ночь.
Неврологический статус: Сходящееся косоглазие. Мышечный тонус повышен по спастическому типу, сухожильные рефлексы высокие — S > D, патологические стопные симптомы. Гипотрофия мышц левых конечностей, больше в ноге. Плосковальгусная установка левой стопы, ретракция в левом голеностопном суставе. Ретракция в левом локтевом суставе с ротацией предплечья. Расторможен, эйфоричен, снижено чувство дистанции. Речь фразовая.
Проведено обследование: биохимический анализ крови, ЭКГ, УЗИ внутренних органов без патологии. По данным МРТ головного мозга: локальные атрофические, мультифокальные мелкоочаговые кистозно-глиозные изменения правой лобно-теменной области. Психолог: интеллектуальное развитие не соответствует возрастному уровню. Окулист: патологии не выявлено. ЭЭГ: умеренные функционально-органические изменения. Региональная эпилептиформная активность в правой центральной области, по морфологии схожая с роландической. Видео-ЭЭГ-мониторинг сна: выраженные изменения БЭА, выражающейся в задержке формирования основных ритмов, стойкой эпилептиформной активности в лобных отделах правого полушария головного мозга, усиливающейся во сне. Иктальная ЭЭГ характерна для пароксизмов эпилептической природы; вероятная локализация фокуса эпиактивности в дополнительной моторной зоне лобной доли правого полушария. Нельзя полностью исключить наличие ночной пароксизмальной дистонии.
Получает терапию: депакин хроно — 500 мг/сут, финлепсин ретард — 200 мг/сут, мидокалм 0,05 — 1 таблетка 3 раза в сутки, фенибут 0,25 — 1/2 таблетки на ночь, наком — 1/4 таблетки утром. На фоне лечения эпилептических приступов не наблюдается, сохраняются редкие дистонические атаки во сне.
Генетика пароксизмальных дискинезий представлена в таблице 2. Следует заметить, что в нашей стране генетическая диагностика пароксизмальных дискинезий пока не проводится. Истинная частота пароксизмальных дискинезий в общей популяции неизвестна, поскольку обычно эти состояния ошибочно диагностируются как другие заболевания. Упоминавшиеся выше пациенты долгое время наблюдались с диагнозом эпилепсии (та или иная форма) и длительно получали антиэпилептическую терапию, которая оказывала разное действие на атаки гиперкинезов. Этиология вторичных пароксизмальных дискинезий включает рассеянный склероз, перинатальные поражения головного мозга и ДЦП, психогении, черепно-мозговые травмы, энцефалиты, опухоли, патологию щитовидной железы и болезнь Фара, дисгенезии головного мозга, инсульты, сахарный диабет и др. В данном случае представлены истории болезни двоих детей с ДЦП (пароксизмальная дискинезия является симптоматической) [6].
Ведущим звеном патогенеза исследователи признают дисфункцию базальных ганглиев. У некоторых пациентов при проведении позитронно-эмиссионной томографии (ПЭТ) и однофотонной эмиссионной компьютерной томографии (СПЕКТ) выявлены изменения в обмене катехоламинов. Кроме того, у ряда пациентов эффективными оказались препараты леводопы (предшественник нейромедиаторов дофаминового ряда) или нейролептики (антагонисты дофамина). В исследованиях на животных при ПГД повышение активности главного тормозного нейромедиатора — γ-аминомасляной кислоты (ГАМК) уменьшает тяжесть и частоту атак. Известно, что ГАМКергическим действием обладают антиконвульсанты — фенобарбитал, вальпроаты, бензодиазепины. При клинических наблюдениях у пациентов с ПГД отмечалась эффективность антиконвульсантов, а в случаях ПНКД течение заболевания смягчалось при применении лишь вальпроатов и фенобарбитала. Это доказывает, что в основе форм пароксизмальных дискинезий лежат разные механизмы. Однако более детальных исследований их патогенеза до настоящего времени не проводилось.
Морфологический субстрат пароксизмальных дискинезий неизвестен. Поскольку участок 2 хромосомы (2q33–q35), поврежденный при пароксизмальных дискинезиях, содержит гены каналов мембраны, существует точка зрения, что они являются «каналопатиями», т. е. возникают в результате аномальной работы электрических каналов цитоплазматической мембраны [2]. Нарушение работы каналов приводят к изменениям концентраций ионов в клетке и внеклеточном пространстве. Мутации тех же генов обнаружены при некоторых других каналопатиях, например семейном гипер- или гипокалиемическом параличе, пароксизмальной атаксии.
Диагностика пароксизмальных дискинезий основана на клинических симптомах (продолжительность и частота атак, «провоцирующие» факторы; наличие сходных проявлений у родственников). Следует провести лабораторные анализы, ЭЭГ (желательно — мониторинг во время атаки гиперкинезов), МРТ — все это позволит исключить болезни со сходной клиникой, а также выявить текущие заболевания, при которых пароксизмальная дискинезия является вторичной. Для идиопатических пароксизмальных дискинезий не характерны изменения на МРТ или компьютерной томограмме. Остается открытым вопрос, являются ли пароксизмальные дискинезии эпилепсиями. Ряд исследователей относят пароксизмальные дискинезии к эпилепсиям, основываясь на клинических проявлениях (пароксизмальность, предвестники, эффект от антиэпилептических препаратов). Другие авторы указывают на то, что в период атаки нет изменений на ЭЭГ, отсутствуют изменения сознания и поведения после приступа. Описаны случаи регистрации у пациентов с пароксизмальными дискинезиями неспецифических патологических паттернов на ЭЭГ и «сосуществования» у одного пациента пароксизмальной дискинезии и эпилепсии [2, 4]. Особенно трудно отличить ночные атаки ПГК от ночных лобных приступов при эпилепсии, тем более что в обоих случаях эффективны антиэпилептические препараты. Единственный метод, позволяющий правильно установить диагноз, — ночной видео-ЭЭГ-мониторинг, который во время ночных атак гипногенной пароксизмальной дистонии не выявляет эпилептиформной активности [1]. Алгоритм дифференциальной диагностики представлен на рисунке. С нашей точки зрения, пароксизмальные дискинезии не являются эпилепсиями по нескольким причинам:
Лечение ПД очень сложно. Единые рекомендации по терапии не разработаны. По нашему мнению, трудность подбора терапии связана с выраженными колебаниями эндогенных катехоламинов в приступном и межприступном периодах. Возможно, поэтому для купирования атак гиперкинезов (в остром состоянии) лучше назначать нейролептики (препарат выбора — тиапридал), а для профилактики атак — комбинацию накома и антиэпилептического препарата (депакин/фризиум).
В то же время данную схему нельзя рассматривать в качестве «жесткой» рекомендации для каждого пациента, так как в некоторых случаях эффективны и другие препараты (финлепсин, пропранолол, высокие дозы пирацетама и др.). Безусловно, следует учитывать и вероятность возникновения побочных эффектов в результате лечения, поскольку атаки регрессируют с возрастом.
Литература
М. Ю. Бобылова
Е. С. Ильина, кандидат медицинских наук
С. В. Пилия
М. Б. Миронов, кандидат медицинских наук
И. А. Васильева
А. А. Холин, кандидат медицинских наук
С. В. Михайлова, кандидат медицинских наук
А. С. Петрухин, доктор медицинских наук, профессор
РГМУ, РДКБ Росздрава РФ, Москва



