Период полувыведения (полужизни)
Период полувыведения [ править | править код ]
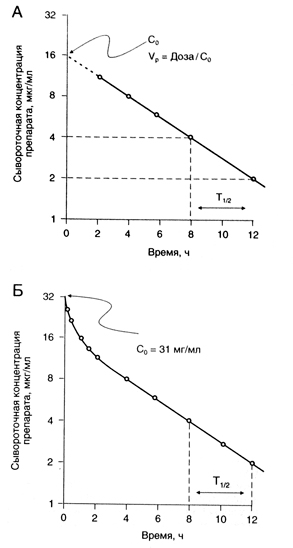
Период полувыведения (Т1/2) — это время, за которое сывороточная концентрация вещества (или его общее содержание в организме) снижается вдвое. В рамках однокамерной модели (рис. 1.4, А) определить Т1/2 очень просто. Полученное значение используют затем для расчета дозы. Однако для многих лекарственных средств приходится использовать многокамерную модель, так как динамика их сывороточной концентрации описывается несколькими экспоненциальными функциями (рис. 1.4, Б). В таких случаях рассчитывают несколько значений Т1/2.
Раньше Т1/2 рассчитывали по участку фармакокинетической кривой, отражающему стадию равновесия (стадию элиминации). С появлением более чувствительных методов измерения концентрации веществ в крови оказалось, что конечный Т1/2 гораздо больше начального. Например, для гентамицина конечный Т1/2 равен 53 ч, тогда как в Приложении II приведен T1/2 2—3 ч. Чрезвычайно длительный конечный Т1/2 индометацина (120 ч) обусловлен, вероятно, активным кишечно-печеночным кругооборотом препарата (в Приложении II приведен Т 1/2 2,4 ч). Клиническая значимость Т1/2 для того или иного периода зависит от того, какая доля вещества выводится из организма и каков объем распределения в этот период, а также от того, какой из показателей — сывороточная концентрация препарата или его общее содержание в организме — лучше коррелирует с фармакологическими эффектами. В Приложении II приведены величины Т1/2, имеющие наибольшее практическое значение.
В прошлом изменение фармакокинетики лекарственных средств при разных патологических состояниях оценивали только на основании Т1/2. В настоящее время общепризнано, что Т1/2 зависит от клиренса и объема рас- пределения вещества. В стационарном состоянии зависимость между Т1/2, клиренсом и объемом распределения приблизительно описывается следующим уравнением:
T1/2 = 0.693 x Vc / Cl(1.12)
Клиренс характеризует способность организма элиминировать вещество, поэтому при снижении этого показателя вследствие какого-либо заболевания Т1/2 должен увеличиваться. Но это справедливо лишь в том случае, если не меняется объем распределения вещества. Например, с возрастом Т1/2 диазепама увеличивается, но не за счет уменьшения клиренса, а вследствие увеличения объема распределения (Klotzetal., 1975). На клиренс и объем распределения влияет степень связывания вещества с белками плазмы и тканей, так что предсказать изменение Т1/2 при том или ином патологическом состоянии не всегда возможно. При остром вирусном гепатите Т1/2 толбутамида уменьшается, а не увеличивается, как это можно было бы ожидать, из-за снижения степени связывания препарата с белками плазмы и тканей. Объем распределения толбутамида не меняется, а клиренс увеличивается вследствие увеличения сывороточной концентрации свободного препарата (Williams et al., 1977).
По Т1/2 не всегда можно судить об изменении элиминации препарата, зато этот показатель позволяет рассчитать время достижения стационарного состояния (в начале лечения, а также при изменении дозы или частоты введения). Сывороточная концентрация, составляющая примерно 94% средней стационарной, достигается за время, равное 4Т1/2. Кроме того, с помощью Т1/2 можно оценить время, необходимое для полного удаления вещества из организма, и рассчитать интервал между введениями (см. ниже).
Стационарное состояние [ править | править код ]
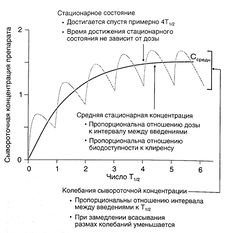
Согласно уравнению 1.1, если вещество вводится путем инфузии с постоянной скоростью, в стационарном состоянии скорость поступления вещества (скорость инфузии) равна скорости его элиминации (произведение клиренса на сывороточную концентрацию препарата — уравнение 1.3). Уравнение 1.1 можно применять и при дробном введении (например, 250 мг каждые 8 ч): в этом случае также устанавливается стационарная сывороточная концентрация препарата, но в промежутках между введениями она колеблется от минимальной до максимальной (рис. 1.5).
Описание к рис. 1.5. Динамика сывороточной концентрации лекарственного средства при дробном введении. Серая кривая описывает накопление препарата при введении с интервалами, равными Т1/2, при условии, что скорость всасывания в 10 раз больше скорости элиминации. При увеличении скорости всасывания максимальная концентрация в стационарном состоянии стремится к 2, а минимальная — к 1. Черная кривая отражает динамику сывороточной концентрации препарата, который вводят в эквивалентной дозе путем инфузии. Обе кривые соответствуют однокамерной фармакокинетической модели. Средняя концентрация в стационарном состоянии вычисляется по уравнению:
Cсредн=F x Доза / (Cl x T)
Это уравнение можно получить путем замены в уравнении 1.1 скорости поступления вещества на выражение F х Доза / Т. Ссредн соответствует концентрации препарата в стационарном состоянии при введении путем инфузии.
Что такое период полувыведения лекарственного препарата
Такие процессы, как всасывание и выведение, обладают экспоненциальными характеристиками. В отношении всасывания это следует из простого факта: количество препарата, перемещающегося за единицу времени, зависит от разности концентраций (градиента) на границе двух тканей (закон Фика).
В процессе всасывания из пищеварительного тракта содержимое кишечника и кровь представляют собой ткани с изначально высокой и низкой концентрациями соответственно. При выведении лекарственного вещества через почки экскреция часто зависит от скорости клубочковой фильтрации, т. е. от количества препарата, попавшего в первичную мочу.
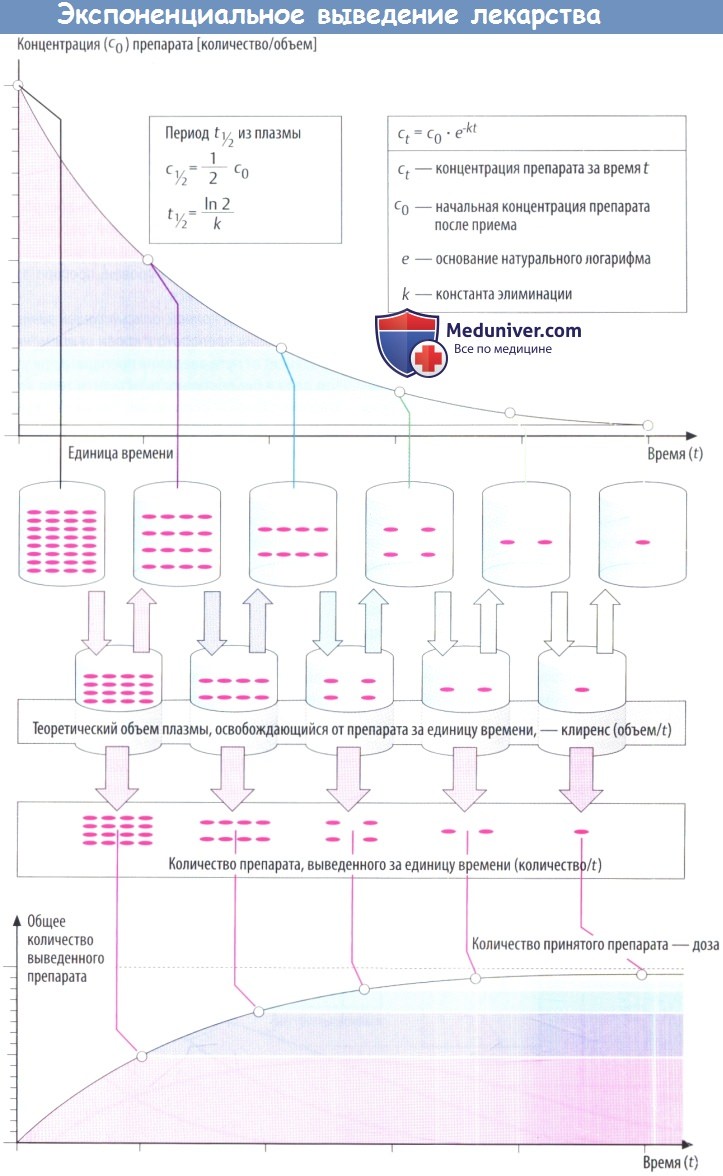
По мере снижения концентрации в крови количество лекарственного вещества, экскретируемого через почки за единицу времени, уменьшается. В результате происходит экспоненциальное снижение, показанное на рисунке ниже. Время экспоненциальногоснижения — постоянный интервал, в течение которого концентрация снижается в 2 раза.
Этот интервал представляет собой период полувыведения (t1/2) и связан с константой скорости элиминации (k) уравнением: t1/2 = (ln2)/k. Эти два параметра вместе с исходной концентрацией (с0) описывают скорость реакции первого порядка (экспоненциальную).
Поскольку эта скорость постоянная, она дает возможность вычислить объем плазмы, освобожденной от лекарственного вещества, учитывая, что оставшееся количество не распределено равномерно в общем объеме плазмы (условие, невозможное в реальности). Теоретический объем плазмы, освобождающейся от лекарственного вещества за единицу времени, называется клиренсом.
В зависимости от того, снижается концентрация в плазме в результате экскреции с мочой либо в результате разрушения в процессе метаболизма, клиренс называют почечным или печеночным. Почечный и печеночный клиренсы суммируются, образуя общий клиренс (Cltot) в случае, если препараты выводятся в неизмененном виде через почки и подвергаются биотрансформации в печени.
Cltot представляет собой сумму всех процессов, участвующих в выведении; он связан с периодом полувыведения (t1/2) и объемом распределения препарата (Vapp) формулой:
Чем меньше объем распределения и чем больше общий клиренс, тем короче период полувыведения.
Для препаратов, выводимых почками в неизмененном виде, t1/2 можно вычислить на основании кумулятивной экскреции с мочой; итоговое общее количество выведенного препарата соответствует количеству всосавшегося препарата.
Печеночная элиминация происходит по экспоненте, т. к. ферменты, катализирующие реакции метаболизма, действуют в квазилинейной области своей кривой активности концентрации; следовательно, количество вещества, подвергшегося метаболизму за единицу времени, уменьшается параллельно снижению концентрации в крови.
Самое известное исключение из экспоненциального закона — выведение алкоголя (этанола), которое происходит по линейному закону (кинетика нулевого порядка), во всяком случае, при концентрации в крови менее 0,02%. Это происходит потому, что лимитирующий скорость фермент алкогольдегидрогеназа достигает полунасыщения при очень низких концентрациях вещества — примерно 80 мг/л (0,008%).
Таким образом, при концентрации этанола в крови на уровне примерно 0,02% скорость реакции выходит на плато, при концентрациях выше этого уровня количество лекарственного вещества, выведенного за единицу времени, остается постоянным.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Период выведения и время полураспада лекарств

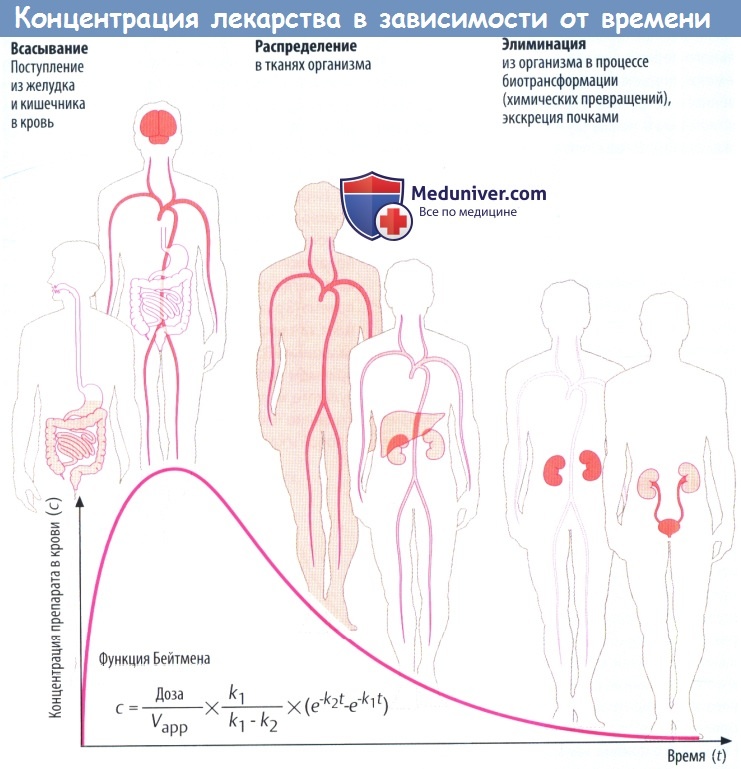
После всасывания в кровь лекарственные средства (ЛС) неравномерно распределяются в органах и тканях организма. Существенно влияют на распространение веществ биобарьеры. К ним относятся стенка капилляров, цитоплазматический, гематоэнцефалический (ГЭБ) и плацентарный барьеры.
Биологические барьеры организма
Большинство препаратов легко преодолевает стенку капилляров. Одни средства проникают через поры путем фильтрации, другие проникают через капиллярную стенку путем диффузии. Некоторые гидрофильные соединения преодолевают капиллярную стенку с помощью транспортных систем.
Выведение лекарств из организма
ЛС и их метаболиты выводятся из организма разнообразными путями: с мочой, калом, желчью, секретом потовых, сальных и бронхиальных желез, молоком матери, воздухом, выдыхаемым воздухом.
Базовую роль в экскреции лекарств играют почки. На выведение лекарств влияют фильтрация, канальцевая реабсорбция и секреция. Фильтрации в клубочках нефрона испытывают вода, глюкоза, аминокислоты, белки с молекулярной массой до 60000 и некоторые другие соединения. Не фильтруются фракции препаратов, связанные с белками плазмы. Скорость фильтрации зависит от интенсивности кровообращения в почках.
В случаях, когда почечный кровоток нарушен (шок, гломерулонефрит и др.), фильтрация существенно уменьшается.
Выделение лекарств с мочой
Активная секреция лекарственных средств происходит в проксимальных отделах нефрона. Секреция из крови через канальцевый эпителий в первичную мочу происходит с затратой энергии с участием специальных транспортных систем. Секретироваться могут как свободные, так и связанные с белками лекарственные средства. Реабсорбция лекарств происходит в дистальных отделах канальцев. Поскольку пассивная реабсорбция происходит через липидные мембраны канальцевого эпителия, то становится очевидным, что лучше реабсорбируются недиссоциированные липофильные молекулы слабых кислот и щелочей, а также нейтральные соединения. Степень реабсорбции зависит от рН мочи. Так, при кислых рН мочи слабые кислоты (барбитураты, бензодиазепины, сульфаниламиды) мало диссоциированные и легко реабсорбируются в кровь.
Выделение лекарств с калом
С калом выводятся из организма препараты, которые плохо всасываются в желудочно-кишечном тракте. Такие препараты используют преимущественно для воздействия на микрофлору кишечника или как слабительные средства.
Некоторые препараты (тетрациклин, пенициллины и др.) выделяются с желчью в тонкий кишечник, откуда они могут выводиться с калом или повторно всасываться, а затем снова выделяться в кишечник (так называемая циркуляция по энтеропеченочную кругу).
Другие способы выведения лекарств из организма
Период полувыведения
Необходимо отметить, что с увеличением дозы препарата выведение его из организма снижается и соответственно возрастает период полувыведения.
Кроме того, для количественной характеристики скорости вывода вещества из организма используют термин «клиренс» (очищение). Он отражает скорость очистки плазмы крови от вещества (например, 10 мл / мин). Различают общий, почечный и печеночный клиренс.
Большинство лекарственных средств несут в организм метаболические изменения. Этот процесс называется биотрансформацией. Суть метаболических превращений заключается в том, чтобы чужеродное, опасное для организма средство превратилось в соединение, которое может быть легко выведено с мочой, желчью или потом. Такие полярные метаболиты плохо растворяются в липидах и имеют низкую способность взаимодействовать с белками плазмы крови и тканей. Метаболиты, как правило, плохо проникают через биологические мембраны и не испытывают реабсорбции в почках и кишечнике.
Метаболизм лекарств в организме
Метаболизм лекарственных средств происходит преимущественно в микросомальном аппарате печени. Некоторые метаболические преобразования определенных лекарств могут происходить в кишечнике, легких, коже и плазме крови. Лишь некоторые препараты выводятся из организма в неизмененном виде.
Известны два базовых вида метаболизма ЛС:
Восстановление является более редким путем метаболизма лекарств. Реакции восстановления катализируют такие ферментные системы, как нитро- и азоредуктазы и др.
Процессы обезвреживания лекарств существенно замедляются у больных с патологией печени (цирроз, острые и хронические гепатиты и др.). Это приводит к росту продолжительности действия препаратов, развития явлений передозировки.
Некоторые препараты могут подавлять микросомальные ферменты печени (левомицетин, бутадион и др.) или немикросомальные ферменты (антихолинэстеразные средства, ингибиторы МАО и др.). В таких случаях действие лекарств, метаболизм которых происходит при участии соответствующих ферментов, увеличивается. В то же время существуют соединения (фенобарбитал и др.), которые повышают (индуцируют) скорость синтеза микросомальных ферментов.
Что такое период полувыведения лекарственного препарата
В распределении анестетика важную роль играет также связывание с белками плазмы, наибольшее значение из которых имеют альбумины. Связанная с белками часть препарата образует депо и находится в равновесии с растворенной в плазме частью, но лишь растворенная часть препарата (не связанная с белками плазмы) распределяется в тканях и оказывает фармакологическое действие. Различные прераты при одновременном введении вместе конкурируют за места связывания с белками плазмы.
В результате концентрация свободной фракции отдельных препаратов в плазме может возрасти, что проявляется признаками передозировки. Аналогичный эффект может вызвать уменьшение белково-связывающей емкости плазмы при заболеваниях печени и почек, а также алиментарной белковой недостаточности. В этих случаях дозу анестетика можно уменьшить. Еше большее практическое значение имеет зависимость связывания препарата с белками плазмы от скорости его введения.
При быстром введение препарата свободная его фракция (т.е. экологически активная часть) увеличиваются. Во избежание острой передозировки внутривенные анестетики вводят можно и с учетом достигаемого эффекта.|
Элиминация. Внутривенные анестетики подвергаются биотрансформации. Отчасти метаболизируются или инактивируются в печени и выводятся с желчью, почками (гепаторенальный клиренс). Лишь небольшая часть препаратов выдится из организма в неизмененном виде. Метаболизация с участием фермнтов более длительный процесс, чем вывод через легкие. Поэтому время элиминации даже современных внутривенных аанестетиков короткого действия больше ингаляционных анестетиков.
Период полувыведення. При системном применении фармакологических препаратов различают три объема (пространства, камеры) распределения: 1. Плазма крови, составляющая 4 объема тела (центральный объем).
2. Интерстициальное пространство (15%).
3. Внутриклеточный объем (40%). Поскольку эндотелий большинства органов содержит межклеточные поры или фенестрирован, то проникновение веществ через него происходит относительно беспрепятственно и зависит только от размера молекул. В связи с этим представляется привлекательной концепция, согласной которой интерстициальное пространство и плазма с точки зрения фармакокинетики рассматриваются как единое (внеклеточное) пространство.
Для активного проникновения внутривенного анестетика решающее значение имеет скорость, с которой анестетик диффундирует из центрального объема (плазма крови) в более глубокие пространства головного мозга. Математически этот процесс можно описать уравнением, рассчитав равновесный период полувыведения 11/2 keO. Он позволяет судить о начале действия препарата. Время, в течение которого происходит начальное распределение препарата по всему организму, обозначают как период полураспределения (tl/2a).
После завершения распределения между концентрациями препарата в отдельных пространствах устанавливается устойчивое равновесие (steady state). Дальнейшая динамика концентрации определяется в первую очередь процессом элиминации анестетика (клиренс плазмы). Время, необходимое для уменьшения концентрации вещества в плазме до половины исходного уровня, называют периодом полувыведения (tl/2p). Снижение концентрации, как правило, описывается логарифмической зависимостью.
Период полувыведения вещества не следует отождествлять с длительностью его действия (см. выше)! Чтобы рассчитать элиминацию при непрерывном поступлении препарата (ТВА, ИУЦК), основываются на контекстно-чувствительном периоде полувыведения. Под контекстно-чувствительным периодом полувыведения понимают время, за которое концентрация анестетика в плазме крови после прекращения внутривенной его инфузии уменьшается на 50%. Элиминационный период полувыведения анестетика определяют исходя из его плазменного клиренса и объема распределения. Этот период тем короче, чем больше клиренс и чем меньше объем распределения.
Возможности клинического применения левофлоксацина
Антибактериальные препараты из группы фторхинолонов занимают одно из ведущих мест в лечении различных бактериальных инфекций, в том числе в амбулаторных условиях. Однако столь популярные в настоящее время ципрофлоксацин, офлоксацин, ломефлоксацин
Антибактериальные препараты из группы фторхинолонов занимают одно из ведущих мест в лечении различных бактериальных инфекций, в том числе в амбулаторных условиях. Однако столь популярные в настоящее время ципрофлоксацин, офлоксацин, ломефлоксацин, пефлоксацин обладают высокой активностью в отношении грамотрицательных возбудителей, умеренной активностью против атипичных возбудителей и малоактивны в отношении пневмококков и стрептококков, что значительно ограничивает их применение, особенно при респираторных инфекциях.
В последнее десятилетие в клиническую практику стали входить новые препараты из этой группы — т. наз. новые фторхинолоны, которые сохраняют высокую активность против грамотрицательных возбудителей, свойственную их предшественникам, и при этом значительно более активны против грамположительных и атипичных микроорганизмов. Одним из таких препаратов является левофлоксацин (таваник). По химической структуре он представляет собой левовращающий изомер офлоксацина. Широкий спектр антибактериальной активности, высокая безопасность, удобные фармакокинетические свойства обусловливают возможность его широкого применения при различных инфекциях.
Механизм действия
Левофлоксацин обладает быстрым бактерицидным действием, поскольку проникает внутрь микробной клетки и подавляет, так же как и фторхинолоны первого поколения, ДНК-гиразу (топоизомеразу II) бактерий, что нарушает процесс образования бактериальной ДНК. Ферменты клеток человека не чувствительны к фторхинолонам, и последние не оказывают токсического действия на клетки макроорганизма. В отличие от препаратов прежнего поколения новые фторхинолоны ингибируют не только ДНК-гиразу, но и второй фермент, ответственный за синтез ДНК, — топоизомеразу IV, выделенную у некоторых микроорганизмов, прежде всего грамположительных. Считается, что именно воздействием на этот фермент объясняется высокая антипневмококковая и антистафилококковая активность новых фторхинолонов.
Левофлоксацин обладает клинически значимым дозозависимым постантибиотическим эффектом, достоверно более длительным по сравнению с ципрофлоксацином, а также длительным (2-3 часа) субингибирующим действием.
Под действием левофлоксацина отмечено повышение функции полиморфноядерных лимфоцитов у здоровых добровольцев и ВИЧ-инфицированных пациентов. Показано его иммуномодулирующее воздействие на тонзиллярные лимфоциты у больных хроническим тонзиллитом. Полученные данные позволяют говорить не только об антибактериальной активности, но и о синергическом противовоспалительном и антиаллергическом действии левофлоксацина.
Спектр антимикробной активности
Левофлоксацин характеризуется широким антимикробным спектром, включающим грамположительные и грамотрицательные микроорганизмы, в том числе внутриклеточные возбудители (табл. 1).
При сравнении эффективности различных антибактериальных препаратов в отношении возбудителей респираторных инфекций было выявлено, что левофлоксацин в отношении противомикробной активности превосходит остальные препараты. К нему оказались чувствительны все штаммы пневмококка, в том числе пенициллинрезистентные, при сравнительно более низкой чувствительности пневмококков к препаратам сравнения: офлоксацин — 92%, ципрофлоксацин — 82%, кларитромицин — 96%, азитромицин — 94%, амоксициллин/клавуланат — 96%, цефуроксим — 80%. К левофлоксацину оказались также чувствительны все штаммы моракселлы катаралис, гемофильной палочки и метициллин чувствительного золотистого стафилококка, 95% штаммов клебсиеллы пневмонии.
Резистентность
Возможность широкого клинического использования левофлоксацина и других новых фторхинолонов заставляет задуматься об опасности развития резистентности к ним. Хромосомные мутации являются основным механизмом, обеспечивающим устойчивость микробов к фторхинолонам. При этом происходит постепенное накопление мутаций в одном или двух генах и ступенчатое снижение чувствительности. Развитие клинически значимой резистентности пневмококков к левофлоксацину наблюдается после трех мутаций, а следовательно, представляется маловероятным. Это подтверждается и экспериментальными данными: левофлоксацин вызывал спонтанные мутации в 100 раз реже, чем ципрофлоксацин, независимо от чувствительности тестируемых штаммов пневмококка к пенициллину и макролидам. Широкое использование препарата в последние годы в США и Японии не привело к росту резистентности к нему. По данным K. Yamaguchi et al., 1999, чувствительность бактерий к левофлоксацину за пять лет, т. е. с момента начала его широкого применения, не изменилась и превышает 90% как для грамотрицательных, так и для грамположительных возбудителей.
Больший риск развития антибиотикорезистентности связан не с пневмококками, а с грамотрицательными бактериями. В то же время, по некоторым данным, применение левофлоксацина в отделениях интенсивной терапии не сопровождается значимым ростом резистентности грамотрицательной кишечной флоры.
Фармакокинетика
Левофлоксацин хорошо всасывается в желудочно-кишечном тракте. Его биодоступность составляет 99% и более. Так как левофлоксацин почти не подвергается метаболизму в печени, это способствует быстрому достижению максимальной его концентрации в крови (значительно более высокой, чем у ципрофлоксацина). Так, при назначении добровольцам стандартной дозы фторхинолона значения его максимальной концентрации в крови при приеме левофлоксацина составляли 2,48 мкг/мл/70 кг, ципрофлоксацина — 1,2 мкг/мл/70 кг.
После приема разовой дозы левофлоксацина (500 мг) его максимальная концентрация в крови, равная 5,1 ± 0,8 мкг/мл, достигается через 1,3-1,6 часа, при этом бактерицидная активность крови против пневмококков сохраняется до 6,3 часа независимо от их чувствительности к пенициллинам и цефалоспоринам. Более длительное время, до 24 часов, сохраняется бактерицидное действие крови на грамотрицательные бактерии семейства Enterobacteriacae.
Период полувыведения левофлоксацина составляет 6-7,3 часа. Около 87% принятой дозы препарата выделяется с мочой в неизмененном виде в течение последующих 48 часов.
Левофлоксацин быстро проникает в ткани, при этом уровни тканевых концентраций препарата выше, чем в крови. Особенно высокие концентрации устанавливаются в тканях и жидкостях респираторного тракта: альвеолярных макрофагах, слизистой бронхов, бронхиальном секрете. Левофлоксацин также достигает высоких концентраций внутри клеток.
Длительный период полувыведения, достижение высоких тканевых и внутриклеточных концентраций, а также наличие постантибиотического действия — все это позволяет назначать левофлоксацин один раз в сутки.
Лекарственные взаимодействия
Биодоступность левофлоксацина снижается при одновременном приеме с антацидами, сукральфатом, препаратами, содержащими соли железа. Интервал между приемом этих лекарственных средств и левофлоксацина должен составлять не менее 2 часов. Других клинически значимых взаимодействий левофлоксацина выявлено не было.
Клиническая эффективность
Существует много публикаций, посвященных результатам клинических исследований эффективности применения левофлоксацина. Ниже представлены наиболее значимых из них.
В многоцентровом рандомизированном исследовании, включившем 590 пациентов, сравнивалась эффективность и безопасность двух режимов лечения: левофлоксацина в/в и/или перорально в дозе 500 мг в сутки и цефтриаксона в/в 2,0 г в сутки; и/или цефуроксима перорально 500 мг два раза в сутки в комбинации с эритромицином или доксициклином у больных с внебольничной пневмонией. Длительность терапии 7–14 дней. Клиническая эффективность составила 96% в группе левофлоксацина и 90% в группе, получавшей цефалоспорины. Эрадикация возбудителей была достигнута соответственно у 98 и 85% больных. Частота нежелательных действий в группе левофлоксацина составляла 5,8%, а в группе сравнения 8,5%.
В другом большом рандомизированном исследовании сравнивалась эффективность лечения больных с тяжелой пневмонией левофлоксацином 1000 мг в сутки и цефтриаксоном — 4 г в сутки. Первые дни левофлоксацин назначили в/в, затем перорально. Результаты лечения в обеих группах оказались сопоставимы, но в группе цефтриаксона наблюдалась достоверно более частая смена антибиотика в первые дни лечения — из-за недостаточного клинического эффекта.
Сопоставимые результаты были получены и при сравнении групп пациентов, получавших лечение левофлоксацином и коамоксиклавом.
Эффективность монотерапии левофлоксацином изучалась у более чем 1000 пациентов с внебольничной пневмонией. Клиническая и бактериологическая эффективность составляли здесь 94 и 96% соответственно.
Фармакоэкономические исследования показали, что общие затраты на лечение пациентов левофлоксацином и комбинацией цефалоспорина и макролида сопоставимы или даже несколько ниже в группе левофлоксацина.
У пациентов с обострением хронического бронхита сравнивалась эффективность лечения левофлоксацином в дозе 500 мг в сутки перорально и цефуроксимом аксетила внутрь 500 мг два раза в сутки. При этом клиническая и бактериологическая эффективность не различалась в зависимости от групп и составляла 77–97%.
Таким образом, в настоящее время можно считать доказанной высокую эффективность левофлоксацина при респираторных инфекциях нижних дыхательных путей. Результаты проведенных исследований позволили включить левофлоксацин как препарат первого ряда или альтернативный в схему лечения пациентов с внебольничной пневмонией и обострениями хронического бронхита (табл. 2).
В последние годы левофлоксацин стал более широко использоваться и при других инфекционных заболеваниях. Так, появились сообщения, касающиеся его успешного применения у больных с острыми синуситами. Левофлоксацин в 100% случаев активен против наиболее часто встречающихся бактериальных возбудителей этого заболевания; по эффективности он сопоставим с амоксициллином/клавуланатом в больших дозах и значительно превосходит цефалоспорины, котримоксазол, макролиды и доксициклин.
Среди возбудителей урологических инфекций наблюдается рост резистентности к широко используемым антибактериальным препаратам. Так, за период с 1992 по 1996 год отмечено увеличение устойчивости E. Coli и S. saprophyticus к котримоксазолу — на 8–16% и к ампициллину — на 20%. Устойчивость к ципрофлоксацину, нитрофуранам и гентамицину возрасла за этот же период на 2%. Применение левофлоксацина у пациентов с осложненными мочевыми инфекциями в дозе 250 мг в сутки оказалось эффективным у 86,7% пациентов.
Выше приводились фармакокинетические показатели высокого содержания левофлоксацина в тканях. Это наряду с антимикробным спектром препарата послужило основанием для его использования с целью профилактики инфекционных осложнений при эндоскопических методах лечения и диагностики, например при ретроградной холангиопанкреатографии и для периоперационной профилактики в ортопедии.
Применение левофлоксацина в этих ситуациях представляется перспективным и требует дальнейшего изучения.
Безопасность
Левофлоксацин считается одним из самых безопасных антибактериальных препаратов. Однако при его назначении существует ряд ограничений.
У пациентов с нарушением функции печени корригировать дозу препарата не нужно, но нарушение функции почек при снижении клиренса креатинина (менее 50 мл/мин) требует уменьшения дозы препарата. Дополнительный прием левофлоксацина после проведения гемодиализа или амбулаторного перитониального диализа не требуется.
Левофлоксацин не применяется у беременных и кормящих женщин, у детей и подростков. Препарат противопоказан пациентам, у которых в анамнезе имеются нежелательные реакции на лечение фторхинолонами.
У больных пожилого и старческого возраста при приеме левофлоксацина не выявлено повышенного риска развития нежелательных побочных реакций и не требуется коррекции доз.
Контролируемые клинические исследования показали, что побочные реакции при применении левофлоксацина возникают редко и большей частью не являются серьезными. Существует зависимость между дозой препарата и частотой развития НД: при суточной дозе 250 мг их частота не превышает 4,0–4,3%, при дозе 500 мг/ сут. — 5,3–26,9%, при дозе 1000 мг/сут. — 22–28,8%. Наиболее часто наблюдались симптомы желудочно-кишечной диспепсии — тошнота и диарея (1,1–2,8%). При внутривенном введении возможно покраснение места инъекции, иногда наблюдается развитие флебитов (1%).
Дозирование
Левофлоксацин выпускается в двух формах: для внутривенного введения и приема внутрь. Применяется по 250-500 мг один раз в сутки, при тяжелых инфекциях возможно назначение по 500 мг два раза в сутки. При внебольничной пневмонии длительность лечения составляет 10-14 дней, при обострении хронического бронхита — 5-7 дней.



