Решена величайшая проблема биологии
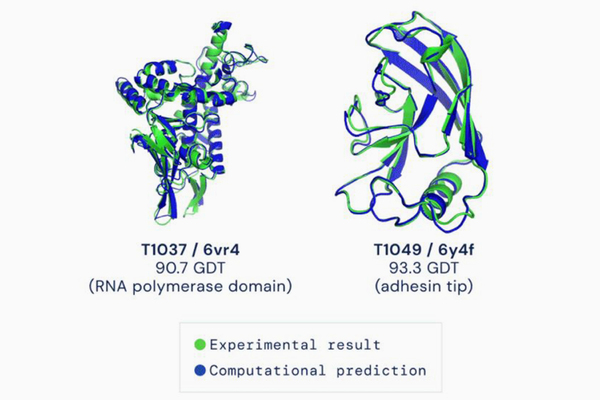
Ученые разработали новую систему искусственного интеллекта DeepMind AlphaFold, способную предсказывать трехмерную структуру белка. Проблема фолдинга (сворачивания) белков была признана биологами всего мира как одна из величайших проблем современной науки, и над ее решением исследователи бились около полувека. О научном прорыве сообщается на сайте DeepMind.


Результаты оценки Critical Assessment of protein Structure Prediction (CASP), проводимая научным сообществом с 1994 года, показали, что AlphaFold обеспечивает беспрецедентную точность в прогнозировании структуры белка. Участники испытаний CASP должны представить систему, способную предсказать, как свернется белок, по последовательности аминокислот, каждая из которых обладает определенными химическими свойствами (например, полярности и гидрофобности). При этом результат прогноза сравнивается с трехмерной структурой, выявленной экспериментальным путем.
Для тестирования используются только те белки, чье сворачивание было изучено лишь недавно, а в некоторых случаях еще не изучено. В последнем варианте нужно ждать, пока трехмерная структура не будет получена для сравнения.
Средняя оценка для AlphaFold составила 92,4 по метрике Global Distance Test (GDT). При этом оценка 90 GDT считается конкурентоспособной среди результатов, полученных экспериментально. Это значит, что искусственный интеллект способен во многих случаях просчитывать трехмерную структуру белков точнее, чем с использованием ряда лабораторных методов.
Трехмерная структура белков тесно связана с их функциями. Однако для каждой аминокислотной последовательности теоретически существует астрономическое число способов сворачивания, но только несколько могут реализовываться и играть важную роль для организма.
Так что же это всё-таки такое, «фолдинг белков»?

В текущей пандемии COVID-19 появилось много проблем, на которые хакеры с удовольствием набрасывались. От лицевых щитков, распечатанных на 3D-принтере и медицинских масок домашнего изготовления до замены полноценного механического аппарата искусственной вентиляции лёгких – этот поток идей вдохновлял и радовал душу. В то же самое время были попытки продвинуться и в другой области: в исследованиях, нацеленных на борьбу непосредственно с самим вирусом.
Судя по всему, наибольший потенциал для остановки текущей пандемии и опережения всех последующих есть у подхода, пытающегося докопаться до самого истока проблемы. Этот подход из разряда «узнай своего врага» исповедует вычислительный проект Folding@Home. Миллионы людей зарегистрировались в проекте и жертвуют часть вычислительных мощностей своих процессоров и GPU, создав таким образом крупнейший [распределённый] суперкомпьютер в истории.
Но для чего конкретно используются все эти экзафлопы? Почему нужно бросать такие вычислительные мощности на фолдинг [укладку] белков? Какая тут работает биохимия, зачем вообще белкам нужно укладываться? Вот краткий обзор фолдинга белков: что это, как он происходит и в чём его важность.
Для начала самое важное: зачем нужны белки?
Белки — жизненно необходимые структуры. Они не только дают строительный материал для клеток, но и служат ферментами-катализаторами практически всех биохимических реакций. Белки, будь они структурными или ферментными, представляют собой длинные цепочки аминокислот, расположенных в определённой последовательности. Функции белков определяются тем, какие аминокислоты расположены в определённых местах белка. Если, к примеру, белку необходимо связываться с положительно заряженной молекулой, место соединения должно быть заполнено отрицательно заряженными аминокислотами.
Чтобы понять, как белки получают структуру, определяющую их функцию, нужно пробежаться по основам молекулярной биологии и информационному потоку в клетке.
Производство, или экспрессия белков начинается с процесса транскрипции. Во время транскрипции двойная спираль ДНК, содержащая в себе генетическую информацию клетки, частично расплетается, давая доступ азотных оснований ДНК ферменту под названием РНК-полимераза. Задача РНК-полимеразы состоит в том, чтобы сделать РНК-копию, или транскрипцию, гена. Эта копия гена под названием матричная РНК (мРНК), представляет собой одинарную молекулу, идеально подходящую для управления внутриклеточными белковыми фабриками, рибосомами, которые занимаются производством, или трансляцией белков.
Рибосомы ведут себя как сборочные приспособления – они захватывают шаблон мРНК и сопоставляют его другим небольшим кусочкам РНК, транспортным РНК (тРНК). У каждой тРНК есть две активные области – секция из трёх оснований под названием антикодон, которая должна совпадать с соответствующими кодонами мРНК, и участок для связывания аминокислоты, специфичной для этого кодона. Во время трансляции молекулы тРНК в рибосоме случайным образом пытаются связаться с мРНК при помощи антикодонов. В случае успеха молекула тРНК присоединяет свою аминокислоту к предыдущей, формируя очередное звено в цепочке аминокислот, закодированной мРНК.
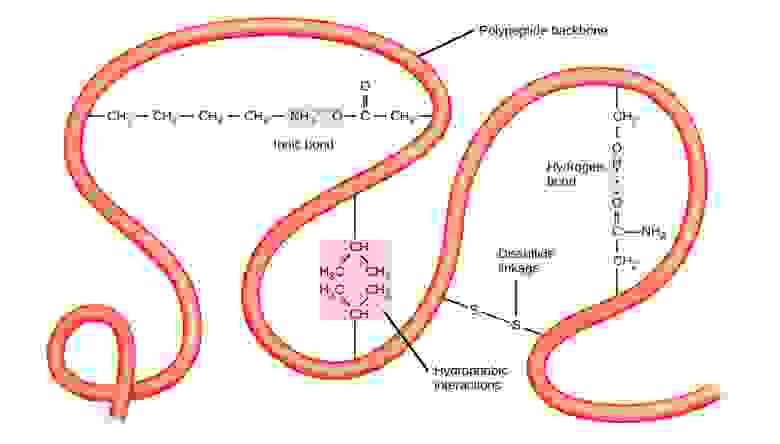
Эта последовательность аминокислот является первым уровнем структурной иерархии белка, поэтому и называется его первичной структурой. Вся трёхмерная структура белка и его функции напрямую происходят от первичной структуры, и зависят от различных свойств каждой из аминокислот и их взаимодействия между собой. Не будь этих химических свойств и взаимодействий аминокислот, полипептиды так и оставались бы линейными последовательностями без трёхмерной структуры. Это можно увидеть каждый раз во время готовки еды – в этом процессе происходит тепловая денатурация трёхмерной структуры белков.
Дальнодействующие связи частей белков
Следующему уровню трёхмерной структуры, выходящему за рамки первичной, дали хитроумное название вторичной структуры. В неё входят водородные связи между аминокислотами относительно близкого действия. Основная суть этих стабилизирующих взаимодействий сводится к двум вещам: альфа-спирали и бета-листу. Альфа-спираль образует туго скрученный участок полипептида, а бета-лист – гладкую и широкую область. У обоих образований есть как структурные, так и функциональные свойства, зависящие от характеристик составляющих их аминокислот. К примеру, если альфа-спираль в основном состоит из гидрофильных аминокислот, как аргинин или лизин, то она, скорее всего, будет участвовать в водных реакциях.

Альфа-спирали и бета-листы в белках. Водородные связи формируются во время экспрессии белка.
Эти две структуры и их комбинации формируют следующий уровень структуры белка — третичную структуру. В отличие от простых фрагментов вторичной структуры, на третичную структуру в основном влияет гидрофобность. В центрах большинства белков содержатся аминокислоты с высокой гидрофобностью, типа аланина или метионина, и вода исключается оттуда из-за «жирной» природы радикалов. Эти структуры часто появляются в трансмембранных белках, встроенных в двойную липидную мембрану, окружающую клетки. Гидрофобные участки белков остаются термодинамически стабильными внутри жировой части мембраны, а гидрофильные участки белка подвергаются воздействию водной среды с обеих её сторон.
Также стабильность третичных структур обеспечивают дальнодействующие связи между аминокислотами. Классическим примером таких связей служит дисульфидный мостик, часто возникающий между двумя радикалами цистеинов. Если в парикмахерской во время процедуры перманентной завивки волос какого-нибудь клиента вы чувствовали запах, немного напоминающей тухлые яйца, то это была частичная денатурация третичной структуры содержащегося в волосах кератина, проходящая посредством уменьшения дисульфидных связей при помощи содержащих серу тиольных смесей.
Третичную структуру стабилизируют дальнодействующие взаимодействия, типа гидрофобности или дисульфидных связей
Дисульфидные связи могут возникать между цистеиновыми радикалами в одной полипептидной цепочке, или между цистеинами из разных полных цепочек. Взаимодействия между разными цепочками формируют четвертичный уровень белковой структуры. Прекрасным примером четвертичной структуры служит гемоглобин у вас в крови. Каждая молекула гемоглобина состоит из четырёх одинаковых глобинов, частей белка, каждый из которых удерживается в определённом положении внутри полипептида дисульфидными мостиками, а также связан с молекулой гема, содержащей железо. Все четыре глобина связаны межмолекулярными дисульфидными мостиками, а вся молекула целиком связывается сразу с несколькими молекулами воздуха, вплоть до четырёх, и способна отпускать их по необходимости.
Моделирование структур в поисках лечения болезни
Полипептидные цепочки начинают укладываться в итоговую форму во время трансляции, когда растущая цепочка выходит из рибосомы – примерно как отрезок проволоки из сплава с эффектом памяти может принимать сложные формы при нагреве. Однако, как всегда в биологии, всё не так просто.
Во многих клетках перед трансляцией транскрибированные гены подвергаются серьёзному редактированию, значительно меняющему основную структуру белка по сравнению с чистой последовательностью оснований гена. При этом трансляционные механизмы часто заручаются помощью молекулярных сопровождающих, белков, временно связывающихся с нарождающейся полипептидной цепочкой, и не дающих ей принимать какую-либо промежуточную форму, из которой они потом не смогут перейти к окончательной.
Это всё к тому, что предсказание окончательной формы белка не является тривиальной задачей. Десятилетиями единственным способом изучения структуры белков были физические методы типа рентгеновской кристаллографии. Только в конце 1960-х биофизические химики начали строить вычислительные модели фолдинга белка, в основном сконцентрировавшись на моделировании вторичной структуры. Этим методам и их потомкам требуются огромные объёмы входных данных в дополнение к первичной структуре – к примеру, таблицы углов связи аминокислот, списки гидрофобности, заряженные состояния и даже сохранение структуры и функционирование на эволюционных временных отрезках – и всё для того, чтобы догадаться, как будет выглядеть окончательный белок.
Сегодняшние вычислительные методы предсказания вторичной структуры, работающие, в частности, в сети Folding@Home, работают примерно с 80% точностью – что довольно неплохо, учитывая сложность задачи. Данные, полученные предсказательными моделями по таким белкам, как белок шипов SARS-CoV-2, будут сопоставлены с данными физического изучения вируса. В итоге можно будет получить точную структуру белка и, возможно, разобраться в том, как вирус прикрепляется к рецепторам ангиотензинпревращающего фермента 2 человека, находящимся в дыхательных путях, ведущих внутрь тела. Если мы сможем разобраться в этой структуре, мы, вероятно, сумеем найти лекарства, блокирующие связывание и предотвращающие инфицирование.
Исследования фолдинга белка лежат в самом сердце нашего понимания такого количества заболеваний и инфекций, что даже когда мы при помощи сети Folding@Home придумаем, как победить COVID-19, за взрывным ростом которого мы наблюдаем в последнее время, эта сеть не будет долго простаивать без работы. Это исследовательский инструмент, отлично подходящий для изучения белковых моделей, лежащих в основе десятков заболеваний, связанных с неправильным фолдингом белков – например, с болезнью Альцгеймера или с разновидностью болезни Крейтцфельдта — Якоба, которую часто некорректно именуют коровьим бешенством. И когда неизбежно появится очередной вирус, мы уже будем готовы снова начать с ним борьбу.
В чём состоит задача фолдинга белков? Краткое пояснение

Белок бактерии Staphylococcus aureus
В конце ноября команда Google DeepMind объявила о том, что её система глубокого обучения AlphaFold достигла небывалых уровней точности в решении задачи фолдинга белков – трудной проблемы из области вычислительной биохимии.
В чём состоит эта проблема и почему её так трудно решить?
Белки – это длинные цепочки аминокислот. Ваша ДНК кодирует эти последовательности, а РНК помогает производить белки согласно этой генетической схеме. Белки синтезируются в виде линейных цепочек, но впоследствии сворачиваются в сложные шарообразные структуры (см. картинку в начале статьи).
Часть цепочки может свернуться в плотную спираль, «α-спираль». Другая часть может согнуться туда и обратно, сформировав широкую плоскую фигуру, «β-лист»:
Сама последовательность аминокислот называется первичной структурой. Упомянутые фигуры называют вторичной структурой.
Сами эти компоненты также складываются, формируя уникальные сложные формы. Это называется третичной структурой:
Фермент, взятый у бактерии Colwellia psychrerythraea
Белок RRM3
Выглядит беспорядочно. Почему же этот спутанный клубок аминокислот так важен?
Структура белка не случайна! Каждый белок сворачивается в определённую, уникальную, и по большей части предсказуемую структуру, что совершенно необходимо для его правильной работы. Благодаря физической форме белок хорошо подходит к структурам, с которыми он может связываться. Имеют значение и другие физические свойства, особенно распределение по белку электрического заряда. На картинке положительный заряд обозначен синим, отрицательный – красным:
Поверхностное распределение заряда на белке-переносчике липидов растений 1 риса посевного
Если белок, по сути, представляет собой самособирающуюся наномашину, то основным предназначением последовательности аминокислот будет производство его уникальной формы, распределение заряда, и всё прочее, что определяет функцию белка. Как именно происходит этот процесс, пока не совсем ясно – сегодня это активная область исследований.
В любом случае, понимание структуры важно для понимания её работы. Однако последовательность ДНК задаёт только первичную структуру белка. Как нам узнать его вторичную и третичную структуры – то есть, точную форму, которую примет этот клубок?
Эту задачу называют задачей фолдинга белков, и к ней есть два базовых подхода: измерение и предсказание.

Дифракционная рентгеновская картина протеазы SARS
Однако эти методы дороги, сложны и времязатратны, а кроме того, работают не со всеми белками. В частности, белки, встроенные в клеточную мембрану – тот же рецептор ангиотензинпревращающий фермент 2 (ACE2), к которому привязывается вирус COVID-19 – складывается в липидном бислое клетки, и его очень сложно кристаллизовать.
Строение клеточной мембраны
Поэтому мы смогли разобрать структуры крохотного процента от секвенированных белков. В универсальной базе данных белков содержится 180 млн последовательностей, а в базе данных трёхмерных структур белков – всего 170 тысяч позиций.
Нам нужен метод получше.
Вспомним, что вторичная и третичная структуры белков в основном являются функцией первичной структуры, известной нам благодаря секвенированию. Что если, вместо того, чтобы измерять структуру белка, мы могли бы её предсказать?
Это задача предсказания структуры белков. Специалисты по вычислительной биохимии работают над ней уже несколько десятилетий.
Как к ней можно подступиться?
Очевидный способ – симулировать физику процесса напрямую. Моделируем силы для каждого атома, учитывая его местоположение, заряд и химические связи. Считаем ускорения и скорости, и пошагово прокручиваем эволюцию системы. Это называется «молекулярной динамикой».

Суперкомпьютер «Антон» компании D. E. Shaw Research

Суперкомпьютер IBM Blue Gene

Онлайн-головоломка Foldit
Поэтому было предпринято уже множество попыток ускорить подобные вычисления. «Антон», суперкомпьютер от D. E. Shaw Research, использует особое оборудование – специальные интегральные схемы. IBM тоже использует био суперкомпьютер Blue Gene. В Стэнфорде запустили проект Folding@Home, использующий распределённые мощности домашних компьютеров. Проект Foldit от UW превратил фолдинг в игру, чтобы дополнить вычисления интуицией человека.
И всё же долгое время ни одна технология не справлялась с предсказанием широкого спектра белковых структур с большой точностью. На проходящих два раза в год соревнованиях CASP, где результаты работы алгоритмов сравниваются со структурами, измеренными экспериментально, первые места получали предсказания с точностью в 30-40%. До недавнего времени:
Медианная точность предсказаний в категории свободного моделирования у лучшей из команд
Как же работает AlphaFold? Она использует несколько глубоких нейросетей, чтобы обучаться разным функциям, связанным с каждым из белков. Одна из ключевых функций – предсказание итоговых расстояний между парами аминокислот. Это приводит алгоритм к итоговой структуре. В одном из вариантов алгоритма (описанном в журналах Nature и Proteins) была выведена потенциальная функция этого предсказания, к которой был применён простейший градиентный спуск, сработавший на удивление хорошо.
Главное преимущество AlphaFold над предыдущими методами – ему не нужно строить предположения касательно структур. Некоторые методы работают, разбивая белки на участки, просчитывая каждый из них, а потом собирая всё обратно. AlphaFold это не нужно.
Судя по всему, в DeepMind считают проблему фолдинга решённой, что мне кажется излишним упрощением, однако в любом случае их прогресс значителен. Эксперты, не связанные с Google, используют такие эпитеты, как «фантастический» и «революционный».
Теперь у генной инженерии есть уже два мощных инструмента, CRISPR и фолдинг белков. Возможно, 2020-е годы станут для биотехнологий такими же, какими 1970-е были для вычислительной техники.
Поздравляем исследователей из DeepMind с этим прорывом!
Проблема фолдинга белка
Проблема фолдинга белка
Из такой цепочки аминокислот в результате ее пространственной укладки может получиться хорошо функционирующий белок. Во многом процесс сворачивания белковой цепи пока неясен и представляет одну из крупнейших проблем современной науки.
Автор
Редакторы
Статья на конкурс «био/мол/текст»: Белки — главные биологические молекулы. Они выполняют множество разнообразных функций: каталитическую, структурную, транспортную, рецепторную и многие другие. Даже всем известная ДНК играет лишь роль «флешки», храня информацию о белках, в то время как белки — сами «файлы». Жизнь на Земле по праву можно назвать белковой. Но так ли много мы знаем о структуре и функционировании этих веществ? До сих пор тайной остается фолдинг белка — процесс пространственной упаковки белковой молекулы, принятия белком строго определенной формы, в которой он выполняет свои функции.

«Био/мол/текст»-2016
Эта работа опубликована в номинации «Свободная тема» конкурса «био/мол/текст»-2016.
Генеральным спонсором конкурса, согласно нашему краудфандингу, стал предприниматель Константин Синюшин, за что ему огромный человеческий респект!
Спонсором приза зрительских симпатий выступила фирма «Атлас».
Спонсор публикации этой статьи — Лев Макаров.
Белки — биополимеры, которые можно сравнить с бусами, где бусинами являются аминокислоты, соединенные между собой пептидными связями (отсюда другое название белков — полипептиды). В клетке белки синтезируются на специальных молекулярных машинах — рибосомах. Выходя из рибосомы, полипептидная цепь сворачивается, и белок принимает определенную конформацию, то есть пространственную структуру (рис. 1). Жизненно важно, чтобы белок присутствовал в организме в определенной форме, то есть конформация должна быть «правильной» (нативной). Процесс сворачивания белка и называется фолдингом (от англ. folding — сворачивание, укладка; отметим, что термин «фолдинг» применим не только к белкам). Самое интересное, что информация о трехмерной структуре «заложена» в самой последовательности аминокислот. Таким образом, белку, чтобы принять нативную структуру, требуется лишь «знать», в какой последовательности и какие аминокислотные остатки в нем присутствуют. Впервые это было доказано в 1961 году Кристианом Анфинсеном на примере бычьей панкреатической рибонуклеазы [1] (рис. 2). Следует сказать, что, помимо белков, чья пространственная структура строго определяется аминокислотной последовательностью, существуют так называемые неструктурированные белки (intrinsically unfolded proteins, IDPs): некоторые фрагменты таких молекул, а иногда и целые молекулы, способны принимать сразу множество возможных конформаций, причем все они энергетически «равноценны», а такие белки довольно часто встречаются в природе и выполняют важные функции [2]. Существует и другой тип фолдинга, происходящий с помощью специальных белков — шаперонов, но о нем чуть позже.
![]()
Рисунок 1. Котрансляционный фолдинг маленького α-спирального домена. Сворачивание полипептидной цепи многих белков начинается уже в рибосоме во время трансляции белка (то есть его синтеза). Созревающий белок выходит из рибосомы через специальный туннель (на рисунке — затемненная область в большой субъединице), который является важным фактором сворачивания цепи [14], [15], причем С-конец цепи (содержащий карбоксильную группу) фиксирован в рибосоме, а N-конец (содержащий аминогруппу) «продвигается» к выходу и «свисает» из него, когда в туннеле накапливается 30–40 аминокислотных остатков [15]. В туннеле могут формироваться компактизированные незрелые структуры, α-спирали, β-шпильки и маленькие α-спиральные домены [14]. Котрансляционный фолдинг проходит в две стадии: сначала несвернутая цепь (U, unfolded) переходит в компактизированное состояние (C, compacted), которая затем приобретает нативную структуру (N, native).
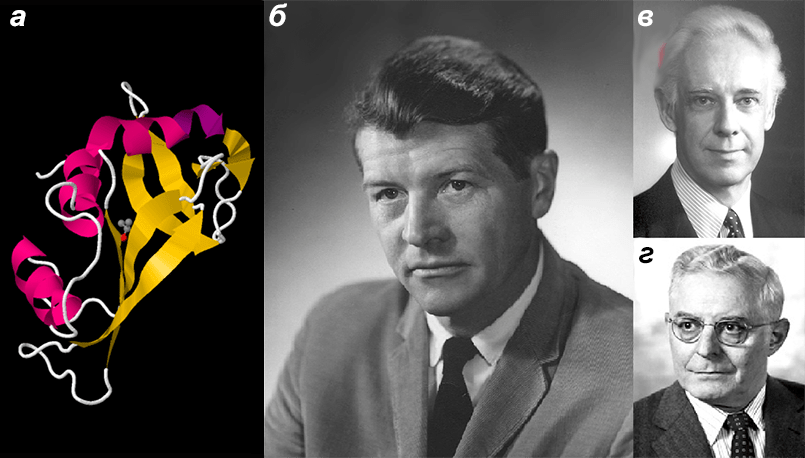
Рисунок 2. Бычья панкреатическая рибонуклеаза и ученые, которые ее изучали. а — Бычья панкреатическая рибонуклеаза. За исследование структуры этого фермента Анфинсен (Anfinsen) (б), Мур (Moore) (в) и Стайн (Stein) (г) получили Нобелевскую премию по химии (1972 г.) [1], [29]. На примере этого белка впервые было показано явление рефолдинга — самопроизвольного формирования третичной структуры после денатурации (то есть разрушения) [1]. Значение белкового фолдинга заключается в том, что он приводит к формированию строго определенной (нативной) структуры белка, в которой он функционирует. Например, в опыте Анфинсена рибонуклеаза в результате рефолдинга восстановила свою ферментативную активность, то есть стала вновь хорошо катализировать биохимическую реакцию. Для того чтобы этот фермент работал, в единый каталитический центр (один кусочек пространства) должны из совершенно разных мест белковой цепи собраться пять аминокислотных остатков: гистидин (12), лизин (41), треонин (47), гистидин (119) и фенилаланин (120) [21].
модель из базы данных PDB (PDB ID 5D6U), портреты ученых с сайта ru.wikipedia.org
Актуальность проблемы
Проблема заключается в том, что человечество со всеми своими вычислительными мощностями и арсеналом экспериментальных данных до сих пор не научилось строить модели, которые бы описывали процесс белкового фолдинга и предсказывать трехмерную структуру белка на основе его первичной структуры (то есть аминокислотной последовательности). Таким образом, полного понимания этого физического процесса до сих пор нет.
Взрывной рост геномных проектов привел к тому, что секвенируется все больше геномов, а соответствующие последовательности ДНК и РНК наполняют базы данных по экспоненте. На рис. 3 изображены рост числа аминокислотных последовательностей, а также рост числа известных белковых структур в период с 1996 по 2007 годы. Хорошо видно, что число известных структур значительно меньше, чем число последовательностей. На момент написания настоящей статьи (август 2016 г.) число последовательностей в базе данных UniParc составляет более 124 миллионов, в то время как количество структур в базе данных PDB (Protein Data Bank) — лишь чуть больше 121 тысячи, что составляет менее 0,1% от всех известных последовательностей, причем разрыв между двумя этими показателями стремительно нарастает и, вероятно, будет расти и дальше. Такое сильное отставание связано с относительной сложностью современных методов определения структур [3]. При этом знать их очень важно. Поэтому вопрос применения вычислительных методов с целью предсказания белковых структур по их последовательностям стоит сейчас остро. В 2005 году авторитетный журнал Science признал проблему фолдинга белка одной из 125 крупнейших проблем современной науки [4].
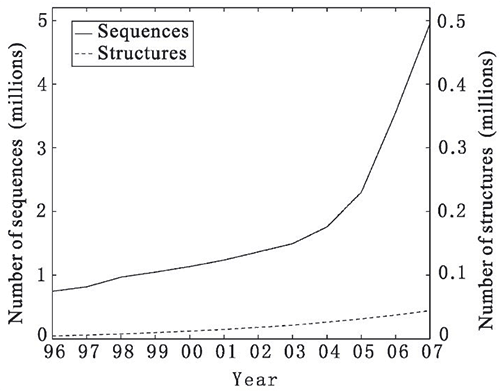
Рисунок 3. Сравнение темпов роста числа известных последовательностей и структур с 1996 по 2007 годы. На горизонтальной оси указываются годы, на левой вертикальной — число последовательностей в миллионах (сплошная линия), на правой вертикальной — число структур в миллионах (пунктирная линия). Четко видно отставание количества известных структур от количества последовательностей. К настоящему моменту разрыв вырос еще сильнее.
После прочтения генома человека стали известны многие человеческие гены и, следовательно, аминокислотные последовательности, кодируемые ими [5]. Однако это не значит, что мы знаем функции всех генов, иначе говоря, мы не знаем функции белков, кодируемых этими генами. Известно, что во многом функции белков можно предсказать по их структуре, хоть и не всегда [6], [7]. Поэтому заветной мечтой является способность предсказывать структуру и, как следствие, функцию белка по самой нуклеотидной последовательности гена.
Что делается для решения проблемы?
Неверно, однако, думать, что мы не знаем совсем ничего. Конечно, накоплено большое количество фактов о фолдинге, известны закономерности этого процесса, разработаны различные методы его моделирования [3], [6–8]. Чтобы отслеживать успехи, достигаемые на пути к решению проблемы фолдинга, был создан международный конкурс по предсказанию пространственной структуры белковых молекул — CASP (Critical Asessement of techniques for protein Structure Prediction), проходящий раз в два года (сейчас соревнование проходит в двенадцатый раз, оно началось в апреле и закончится в декабре 2016 года). В этом состязании исследователи соревнуются, кто лучше предскажет структуру белка по его аминокислотной последовательности, причем конкурс проходит с использованием двойного слепого метода (на момент проведения конкурса структура белка-«загадки» попросту неизвестна; ее определение завершается каждый раз по окончании состязания). Пока что структуры белков-мишеней точно не были предсказаны ни разу.
Существует две группы методов предсказания структуры.
К первой относятся так называемые методы моделирования «с нуля» (ab initio, de novo, есть и другие синонимичные термины), когда модели строятся лишь на основании первичной структуры, без использования сравнительных методов с уже известными структурами, зато с использованием всего накопленного понимания физики сворачивания биополимеров. Фундаментальная значимость этих методов состоит в том, что они помогают понять физико-химические принципы белкового фолдинга, ответить на этот животрепещущий вопрос — почему белок сворачивается так, а не иначе? Однако недостатками этих методов являются очень большая сложность вычисления и невысокая точность [9]. Эти методы требуют упрощений и приближений, а также являются неэффективными для предсказания структур крупных белков. В 2007 году за счет методов моделирования de novo впервые с высокой точностью была определена структура одного из белков бактерии Bacillus halodurans, но белок этот относительно невелик (112 аминокислотных остатков), а для получения точной модели потребовалась мощность более 70 000 персональных компьютеров и суперкомпьютера; кроме того, из 26 полученных моделей точной оказалась лишь одна [10]. Методы молекулярной динамики (МД) позволяют описывать молекулярные события [11] и способны проследить процесс сворачивания белка в нативную структуру: в 2010 году впервые удалось это сделать за счет вычислительной мощности специально созданного суперкомпьютера Anton [12].
Ко второй группе методов относятся методы сопоставительного моделирования. Они основываются на явлении гомологии, то есть общности происхождения объектов (органов, молекул и др.). Таким образом, у «предсказателя» есть возможность сравнивать последовательность белка, структуру которого необходимо смоделировать, с шаблоном, то есть белком, структура которого известна и который предположительно является гомологом, и на основании их схожести строить модель с последующими корректировками (похожие последовательности сворачиваются в похожие структуры). Эти методы сейчас более популярны, так как предсказание структуры белков является важной практической задачей, а к настоящему моменту появились вычислительные средства, базы данных, а также стало известно, что количество возможных вариантов укладок белковых структур ограничено [6], [9] (рис. 4). И пусть эти методы не снимают самой проблемы белкового фолдинга, они способны помочь решать конкретные практические задачи, пока другие бьются над исследованием более фундаментальных вопросов.
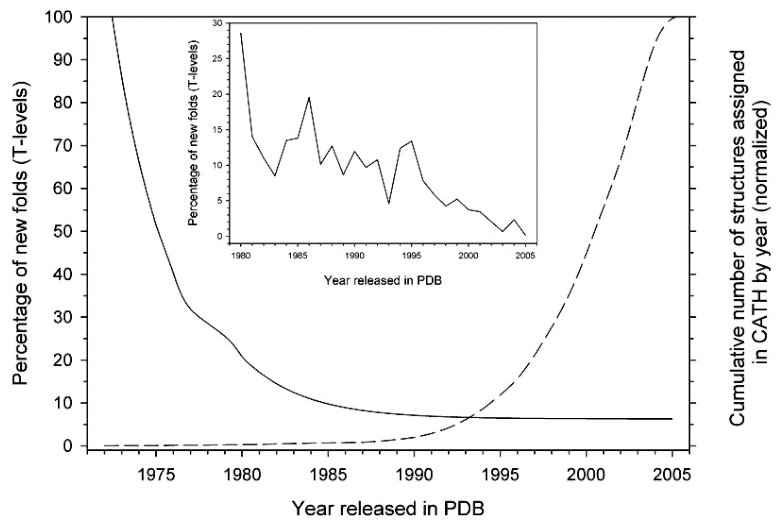
Рисунок 4. Динамика выявления новых типов фолдов (вариантов упаковки). На горизонтальной оси откладывается время (годы), на левой вертикальной оси — доля новых фолдов (более детально — на вкладке) (сплошная линия), а на правой вертикальной оси — общее число структур (пунктирная линия), классифицированных в базе данных CATH. Отметим, что эта база данных занимается структурной классификацией белков, поэтому для нее принципиально знать возможные типы белковых фолдов. Явно видно, что со временем классифицируется все больше и больше белков, но при этом количество вариантов фолдов уменьшается.
Подробнее о применении компьютерных методов для предсказания белковых структур читайте в статье на «биомолекуле»: «Торжество компьютерных методов: предсказание строения белков» [3].
Некоторые закономерности фолдинга белка
Последний этап не происходит при фолдинге неструктурированных белков — IDPs.
Нужно отметить, что для каждой аминокислотной последовательности теоретически можно предположить множество путей, которыми она может идти для достижения нативной конформации. Однако известно, что белок не перебирает все возможные варианты, а движется по одному из возможных путей, определенных для каждой последовательности. Если бы белок пробовал все возможные варианты, то время пути от простой последовательности к нативному состоянию превысило бы время существования Вселенной (парадокс Левинталя)! Конечно, такого не происходит: время принятия белком нативной структуры составляет доли секунды. Это похоже на сборку кубика Рубика: из состояния несобранного кубика к состоянию собранного можно прийти множеством разнообразных путей, однако на соревнованиях по скорости сборки кубика побеждает тот, кто делает это быстрее и эффективнее, то есть выбирает определенный путь. На самом деле найти такой путь — и есть главная задача методов моделирования ab initio (см. выше). Ответ на фундаментальный вопрос фолдинга будет заключаться не просто в способности безошибочно моделировать структуры, а, в первую очередь, в том, чтобы знать и обосновывать путь достижения белком нативного состояния.
Следует подчеркнуть значение котрансляционного фолдинга (рис. 1), о котором говорилось выше, в формировании структуры белка. Отметим, что присутствие рибосомы, на которой синтезируется белок, накладывает серьезные коррективы на процесс сворачивания цепочки. Это всегда нужно иметь в виду при моделировании фолдинга природных белков in vivo. Канал, в котором оказывается растущая цепь, ограничивает ее конформационную изменчивость, а потому далеко не все типы структур могут в ней формироваться [14], [15]. Кроме того, растущая цепочка постоянно проталкивается вперед (на один аминокислотный остаток при каждом акте транспептидации-транслокации, то есть образования новой пептидной связи и последующего продвижения рибосомы), а потому логично будет предположить, что конформация цепи в рибосомном канале обладает такими качествами, как жесткость и векторность, что соответствует свойствам α-спирали [15]. Кроме того, взаимная ориентация аминокислотных остатков в двух центрах внутри рибосомы всегда однотипная (эквивалентная), не зависящая от природы этих остатков, что тоже, по-видимому, способствует формированию α-спиралей [15]. Действительно, α-спирали — наиболее типичный элемент вторичной структуры белков. Они были открыты Лайнусом Полингом (Liunus Pauling) и Робертом Кори (Robert Corey), которые вместе с Уолтером Колтуном (Walter Koltun) предложили новый тип моделей молекул [16].
В то же время, когда N-конец (содержащий аминогруппу) растущей цепи белка выходит из туннеля и погружается в раствор, на него начинают действовать физико-химические условия этой среды, и белок начинает подчиняться их правилам.
Известный молекулярный биолог академик Александр Спирин [15] в этой связи отмечает три различия между фолдингом in vitro и in vivo:
Эти соображения лишний раз доказывают, что биологические вопросы не могут решаться «всухую» за счет применения методов биоинформатики [17]. Даже самые, казалось бы, выверенные компьютерные модели могут оказаться неточны, если они построены без учета факторов, реально действующих в природе.
«Сухая» биология — та, которая использует компьютерные и математические методы, в то время как «мокрая» — это работа с растворами, пипетками, микроскопами и т. д.: «Вычислительное будущее биологии» [18] и «Я б в биоинформатики пошел, пусть меня научат!» [19].
Для решения проблемы фолдинга разработаны так называемые эмпирические потенциалы: парных взаимодействий остатков, водородных связей, торсионных углов, центров масс боковых цепей и многие другие [6], [11]. Например, потенциал сольватации позволяет предсказать, внутри или снаружи белка будет находиться аминокислотный остаток (соответственно заглубленный или экспонированный) в зависимости от его гидрофобности [6], [13]. Известно, что одни аминокислоты «любят» воду (гидрофильные), они будут с большей вероятностью располагаться на поверхности белковой молекулы, а другие — «не любят» (гидрофобные) и «прячутся» в более недоступные для растворителя области молекулы, заслоняясь другими остатками (рис. 5). Гидрофобный эффект имеет большое значение в фолдинге белка.
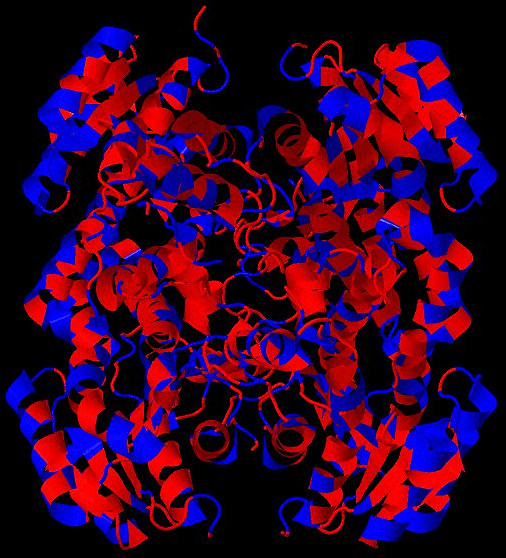
Рисунок 5. Гидрофобность аминокислот влияет на их пространственное распределение (на примере одной из человеческих дегидрогеназ). Гидрофильные аминокислоты показаны синим цветом, гидрофобные — красным. Можно заметить, что гидрофильные остатки стремятся располагаться на открытых для растворителя участках, в то время как гидрофобные — в закрытых областях молекулы.
база данных PDB (PDB ID 5ICS)
Важным аспектом формирования структуры белка на всех этапах является образование связей между радикалами (боковыми цепями) аминокислотных остатков. Они бывают разные: гидрофобные, электростатические и другие [20]. Интересным вариантом является формирование дисульфидных связей («мостиков») за счет взаимодействия атомов серы боковых цепей цистеина. Например, в прославленной рибонуклеазе, за исследование структуры которой была дана Нобелевская премия, таких связей четыре. Однако здесь все не так просто. Если в состав белковой цепи входят два атома серы, принадлежащие цистеину, то легко сказать, что может образоваться один дисульфидный мостик. Но если атомов серы, к примеру, десять и, соответственно, образуются пять SS-связей, то мы не можем однозначно сказать, какие именно атомы серы будут попарно взаимодействовать друг с другом (а белок может). Согласно расчетам Томаса Крейтона (Thomas Creighton), если в белке 5 дисульфидных связей, число возможных комбинаций составляет уже 945, если таких связей 10, то число вариантов составляет 654 729 075, а при 25 дисульфидных связях это число превышает 5 квадриллионов квадриллионов (более 5,8 × 10 30 ) [21]. А в белке реализуется лишь один вариант, и притом всегда один и тот же! Следует тем не менее отметить, что это справедливо для самоорганизации белков in vitro («в пробирке», «в стекле», то есть в условиях эксперимента, а не в живом организме) в подходящих условиях, а in vivo (в живом организме) самоорганизации дисульфидных связей не происходит. Их образование катализирует специальный фермент — протеиндисульфидизомераза [7], [22] или ПДИ, которая к тому же способна «исправлять» ошибки в случае неправильного образования SS-связи, таким образом корректируя процесс фолдинга [22], [23].
Важно понимать, что процесс формирования окончательной структуры белка не заключается лишь в простом сворачивании цепочки. В клетках белки подвергаются ацетилированию, гликозилированию и многим другим модификациям. Поэтому, например, количество разных аминокислот в белках превышает известные 20 («магическая двадцатка», по образному выражению нобелевского лауреата Фрэнсиса Крика). Кроме того, для формирования сложных (олигомерных) белков необходимо формирование специфических связей между отдельными протомерами (например, в молекуле гемоглобина четыре протомера, то есть отдельно синтезированные цепочки). Для многих белков, особенно ферментов, важным является присоединение простетической группы, то есть небелкового компонента. Могут происходить и другие преобразования [7].
Известны многие другие закономерности белкового фолдинга. Завеса тайны постепенно приподнимается. Однако картина до сих пор далека от целостной. Успехи предсказания структур пока только эпизодические. В связи с этим научное сообщество сделало следующий любопытный шаг: оно привлекло к решению вопроса широкую общественность, создав игру FoldIt [24], [25]. Принять участие в мировом соревновании может любой желающий. Суть игры заключается в том, чтобы свернуть белковую цепочку максимально компактно, то есть привести белковую молекулу в такое состояние, в котором свободного места внутри клубочка как можно меньше — именно в таком виде белки присутствуют в природе (рис. 6). С точки зрения термодинамики, такому состоянию соответствует минимум свободной энергии [7], [26]. Чем более компактная молекула получается, чем меньше полостей и открытых гидрофобных участков, чем больше открытых гидрофильных участков, водородных связей в структурах типа β-листов, чем меньше «столкновений» атомов, тем большее количество баллов игроку начисляется. Таким образом, наибольшее количество баллов получает модель с наименьшей свободной энергией. Большинство игроков FoldIt имеют лишь малую биохимическую подготовку либо не имеют ее вовсе [27]. Игра основана на алгоритмах Rosetta и не является моделированием структур de novo, которое, как верно подмечают авторы, все еще остается исключительно сложной проблемой [27].
![]()
Рисунок 6. Сравнение разных форм представления моделей белковых структур (на примере одной из человеческих трансфераз). а — Форма, наглядно демонстрирующая типы вторичных структур. б — Форма, показывающая реальное расположение атомов молекулы белка в пространстве (Space Fill). Хорошо видно, что молекулы белков сильно компактизированы, между атомами мало свободного пространства.
база данных PDB (PDB ID 5CU6)
Группа игроков FoldIt принимает участие в CASP. Игра уже показала свою эффективность в предсказании структур и даже бóльшую эффективность в сравнении с другими методами, а также решила серьезную научную проблему структуры протеазы вируса иммунодефицита обезьян, которую наука не могла решить на протяжении более чем десятилетия [27].
Говоря о применении разных методов и средств для решения обсуждаемой проблемы, всегда нужно помнить, что не все последовательности могут сворачиваться строго определенным образом. Вероятно, мы, глядя на результаты, к которым пришла эволюция к настоящему времени, видим только те последовательности, которые могут сворачиваться, поскольку они хорошо выполняли свои функции и были поддержаны отбором.
«Гувернантки» для белков — шапероны
Говоря о фолдинге, мы акцентировали внимание на относительной автономности этого процесса: белковая молекула принимает определенную конформацию на основании своей первичной структуры, и происходит это в конкретных (что важно) физико-химических условиях (кислотность, температура, природа растворителя и др.). Тем не менее не должно складываться впечатление, будто бы фолдинг абсолютно независим, особенно для крупных белков. Мы лишь упомянули о ферменте ПДИ, помогающем белку правильно свернуться. Кроме этого фермента, есть и другие (например, ППИ — пептидил-пролил-цис/транс-изомераза [22], [23]). Но ферменты — не единственная группа белков, помогающая правильно сворачиваться другим белкам. Существует еще одна особая группа белков, играющих важную роль в фолдинге. Называются они шаперонами.
Важнейшая группа шаперонов — шаперонины. Интересна их структура: они представляют собой бочонки, составленные из двух колец. Сворачивающийся белок попадает внутрь шаперонина, а «вход» закрывается специальной «шапочкой» либо смыканием краев блоков, из которых состоят кольца [28], чтобы белковая молекула не покинула шаперонин раньше времени (рис. 7). В таком защищенном состоянии белок может окончательно принять нативную конформацию. Пока малопонятны процессы, происходящие внутри бочонков-шаперонинов.
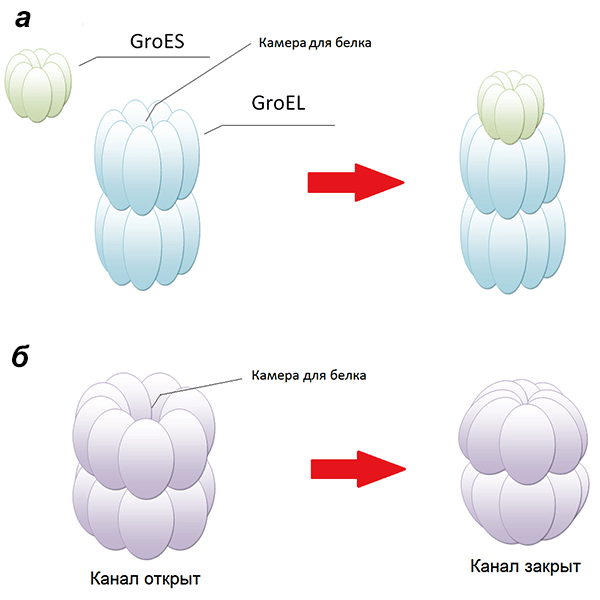
Рисунок 7. Схематическое изображение двух типов шаперонинов — I и II. а — Шаперонины I типа характерны для бактерий (шаперон GroEL имеет структуру бочонка, составленного из двух колец, в каждом — 7 «блоков»; внутри шаперонина — камера, в которой происходит превращение расплавленной глобулы в нативную; бочонок закрывается «крышкой» — GroES); б — Шаперонины II типа, характерные для архей и эукариот (здесь каждое из двух колец состоит из 8 «блоков»; закрытие камеры происходит не за счет присоединения «крышки», а по механизму объектива фотоаппарата [28]).
Нужно сказать, что шапероны не только участвуют в фолдинге созревающих цепей, но и помогают «сломанным» белковым структурам, которые возникли в клетке в результате определенных воздействий, вновь принять правильную конформацию. Наиболее типичная причина таких «поломок» — тепловой шок, то есть поднятие температуры. В связи с этим часто употребляют другие названия шаперонов — белки теплового шока (heat shock proteins, hsp) или белки стресса. Шапероны выполняют другие важные функции в клетке, например, транспорт белков через мембраны и сборку олигомерных белков.
Заключение
Итак, для фолдинга белка строго необходимы следующие условия: первичная структура, конкретные физико-химические условия, а также две группы вспомогательных белков — специфически работающие ферменты и неспецифически работающие шапероны.
Резюмируя, скажем, что белковый фолдинг — одна из центральных проблем современной биофизики. И хотя накоплен большой арсенал данных об этом явлении, до сих пор оно малопонятно, что выражается, в конечном счете, в невозможности предсказания трехмерной структуры на основании аминокислотной последовательности (особенно это касается крупных, в том числе олигомерных, белков). Успехи в этой области, а особенно моделирования de novo, пока можно назвать единичными. Тем не менее понятно, что решение точно есть, ведь в природе белки почти всегда сворачиваются одинаковым, правильным образом. Существует как фундаментальная, так и большая практическая значимость «взлома» кода белкового фолдинга. А это значит, что работы в этой области будут вестись и дальше.



