Что такое синдром ангельмана
Синдром Ангельмана (ранее известный как «синдром счастливой марионетки») включает судорожные движения, беспричинный смех и задержку умственного развития различной, чаще всего серьезной умственной отсталости (Angelman 1965; Horsier и Oliver 2006). Частота в популяции составляет около 1 на 12000 живых новорожденных, с соотношением мужчин и женщин 1:1 (Steffenburg et al., 1996). Исчерпывающее описание синдрома Ангельмана в настоящее время находится в печати (Dan, 2008).
а) Патогенез. Синдром Ангельмана в большинстве случаев вызван делецией 15q11.2-12 хромосомы, сходной, но не идентичной выявляемой у детей с синдромом Прадера-Вилли (Magenis et al., 1987, Knoll et al., 1989) и наследуется по материнской линии (Knoll et al., 1989). Делеция включает ген бета-3-субъединицы ГАМК-рецептора (Saitoh et al., 1994). Среди 60-75% пациентов отмечаются делении или перестройки длинного плеча 15-й хромосомы, делеция всегда располагается на материнской хромосоме.
В небольшом количестве случаев выявляется дисомия отцовской 15-й хромосомы (Malcolm et al., 1991, Prasad и Wagstaff, 1997). Однако не менее чем у 15% пациентов выявляются нормальные хромосомы и отсутствуют признаки дисомии. В некоторых случаях такого рода возможно повторное рождение больных детей среди родственников (Clayton-Smith, 1992). Такие случаи могут быть связаны с доминантной мутацией гена UBE3A (Kishino et al., 1997) на 15q11-13 хромосоме, приводящей к возникновению фенотипа Ангельмана только при передаче по женской линии (Wagstaff et al., 1993).
Различия клинических проявлений зависят от происхождения генетического дефекта (Lossie et al., 2001). Вызванные делециями формы, обычно имеют более выраженный характер, чем формы, вызванные единичной мутацией гена или другими генетическими дефектами. Большинство случаев заболевания носит изолированный характер, но зарегистрированы и семейные случаи.
б) Диагностика. Диагноз подтверждается в случае, если клинический фенотип соответствует положительному FISH-тесту. У младенцев (van Lierde et al., 1990, Yamada и Volpe, 1990) даже при использовании предложенных критериев диагноз зачастую затруднен (Williams et al., 1995). Весьма вероятным диагноз представляется при сочетании задержки умственного развития с проявлениями аутизма, жизнерадостным поведением, атаксией и эпилептическими приступами (часто «минимально» выраженными).
На основании апраксической походки, стереотипиях движений рук и (в некоторых случаях) гипервентиляции возможна ошибочная постановка диагноза синдрома Ретта у девочек. Тем не менее, при раннем (до года) начале припадков, жизнерадостном настроении и дисморфизме вероятность неверной диагностики можно исключить.
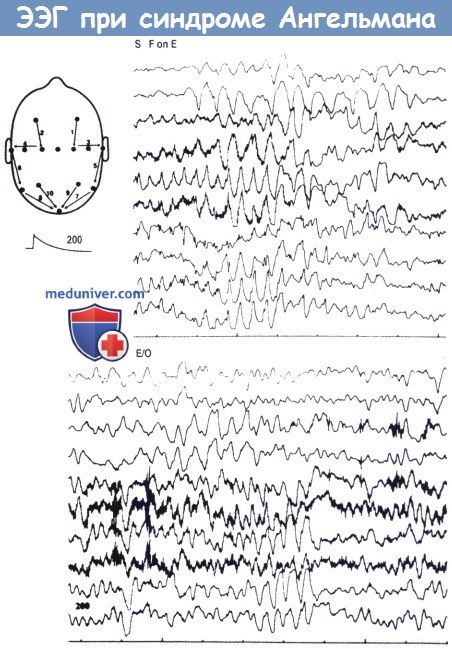 Типичные изменения на ЭЭГ двух детей различного возраста (верхний рисунок —семь лет, нижний рисунок — два года), страдающих синдромом Ангельмана.
Типичные изменения на ЭЭГ двух детей различного возраста (верхний рисунок —семь лет, нижний рисунок — два года), страдающих синдромом Ангельмана.
Следует обратить внимание на калибровку. В обоих случаях отмечается повышенная ритмичная тета-активность, чередующаяся с эпизодами медленного ритма, в частности, в области передней половины двух полушарий.
На верхнем рисунке эпизоды активности с частотой 3-4/с, чередующиеся со слабовыраженными острыми волнами, отмечаются в задней трети головы в случае, когда помощник закрывает пациенту глаза.
S — закрыты, F on Е — «пальцы закрывают глаза», Е/О — глаза открыты.
в) Клинические проявления. Клинические проявления включают серьезную умственную отсталость с тяжелым нарушением речи, атаксию и заметные спастические нарушения движений верхних конечностей, а иногда и туловища; данные двигательные нарушения являются особым типом миоклонуса (Guerrini et al., 1996; Beckung et al., 2004). Немотивированные приступы смеха являются характерным, но не основным проявлением, в то время как жизнерадостное настроение является постоянным признаком (Williams и Frias, 1982).
У 86% больных детей отмечаются приступы, которые часто имеют повторный характер, но длятся недолго и чаще всего являются не тонико-клоническими припадками, а атипичными абсансами или тоническими или атоническими припадками (Dorries et al., 1988; Viani et al., 1995; Laan et al., 1997). На ЭЭГ часто встречаются характерные изменения в виде эпизодов медленных (3 Гц) волн, особенно в задних отделах, часто зазубренных и в отдельных случаях связанных с истинными пиками (Boyd et al, 1988).
На КТ и МРТ каких-либо специфичных изменений не выявляется, но возможно наличие небольшого расширения желудочков и/или околомозгового пространства (Dorries et al., 1988).
Способность к передвижению появляется поздно, часто после 5-6 лет; походка имеет аномальный характер с широко расставленными ногами и атаксическими и апраксическими признаками (Sugimoto et al., 1992).
Дисморфический синдром характеризуется умеренной выраженностью и может быть незаметен в детском возрасте. Прогнатизм обычно развивается только в возрасте нескольких лет.
Большинство случаев заболевания носит изолированный характер, но зарегистрированы и семейные случаи.
Только недавно были опубликованы результаты исследований, подтверждающих мнение о специфическом поведенческом фенотипе данного синдрома, и еще слишком рано делать предположения в данном направлении. Результаты одного из исследований взрослых и молодых пациентов, страдающих синдромом Ангельмана (Clayton-Smith, 1993), свидетельствуют о том, что гиперактивность, часто наиболее выраженная в раннем детском возрасте, в позднем детском возрасте обычно сменяется более контролируемым поведением.
Результаты большинства исследований в настоящее время позволяют предположить, что мутизм может быть постоянным проявлением, но некоторые пациенты могут освоить язык жестов. В одном из центров наблюдался мальчик с сочетанием синдрома Ангельмана и аутизма. У многих детей с синдромом Ангельмана отмечаются заметные аутистические проявления, такие как упрямство, настаивание на одном и том же и увлеченность водой, и в то же время они кажутся счастливыми и общительными, так как им нравится контактировать с другими людьми. Тем не менее, контакт осуществим лишь при выполнении их собственных условий.
г) Исход. Прогноз неблагоприятный, отмечается серьезная умственная отсталость. Овладение коммуникативным языком невозможно (Beckung et al., 2004).
Редактор: Искандер Милевски. Дата публикации: 4.12.2018
Всё о синдроме Ангельмана
В статье расскажем всё о синдроме Ангельмана: определение, историю, причины, симптомы, приведём пример из практики с фото и видео. По каким признакам узнать и как лечить болезнь Ангельмана.
Синдром Ангельмана – это хромосомная патология, локализованная в 15 хромосоме, проявляющаяся выраженной задержкой психо – речевого и моторного развития, расстройством координации, нарушением поведения из группы расстройства аутистического спектра, особенностями эмоциональной сферы («счастливое» выражение лица в сочетание со вспышками смеха, улыбчивость, доверчивость), стереотипиями (взмахи или похлопывания руками), эпилептическими приступами.
Отличительные признаки синдрома Ангельмана
Марионеточная походка, приступы смеха, радостное лицо.
Распространенность синдрома Ангельмана 1 на 10-30 тысяч новорожденных. Но большое количество случаев остаются не диагностированными, а наблюдаются у невролога с задержкой речевого развития, нарушениями поведения, эпилепсией. Мальчики и девочки болеют с одинаковой частотой. При изучении частоты встречаемости среди пациентов с умственной отсталостью выявлено 4,8% больных синдромом Ангельмана.
История
Заболевание названо по имени детского врача из Британии Гарри Ангельмана.
В 1965 году Эйнджелмен (вариант написания) описал трёх мальчиков из разных семей, назвал «Синдром счастливой марионетки» или «happy puppet syndrome», по их ангельскому счастливому внешнему виду и приступам беспричинного смеха, резким движениям в руках, дёрганной марионеточной походкой, тяжелой умственной отсталостью. Аналогия была проведена Эйнджелменом, глядя в музее Castelvecchio Вероны на картину «Мальчик – марионетка» с изображением смеющегося мальчика.
 Карото Джиованни Франческо картина “Мальчик – Марионетка”
Карото Джиованни Франческо картина “Мальчик – Марионетка”
Чарльз Уильямс и Джейми Фриас в 1982 году описали ход болезни и предложили по этическим соображениям термин «синдром счастливой марионетки» заменить по имени описавшего педиатра в Синдром Ангельмана (СА).
Факты сегодняшней жизни
 Colin Farrell с сыном Henry
Colin Farrell с сыном Henry
Ирландский киноактер Колин Фаррелл в 2007 году поделился, что у их сына верифицирован диагноз СА. Как преданный отец Colin James Farrell любит и заботится о сыне, а также оказывает моральную поддержку для подобных семей с детьми – инвалидами. Он участвует в FAST – фонде терапии синдрома Ангельмана. 6 декабря 2013 года в Чикаго был благотворительный Гала вечер Фонда СА, на котором принял участие К. Фаррелл.
Этиологии патогенез
Причина синдрома Ангельмана: в геноме недостаёт части из 3 – 4 миллионов пар оснований ДНК короткого плеча q11—q13 материнской копии пятнадцатой хромосомы.
 Хромосомы человека
Хромосомы человека
Повреждение 15 хромосомы даёт два варианта патологии: при дефекте материнской хромосомы ребенок рождается с синдромом Ангельмана, при дефекте хромосомы отца – с синдромом Прадера – Вилли.
Выявлено четыре генетических варианта появления синдрома Ангельмана:
Риск повторного рождения детей больных этим синдромом при вновь возникшей мутации в одной семье низкий – около 1%. Но в крайне редких случаях риск до 50%, если мутация в центре импритинга.
 Риск повторного рождения детей с синдромом Ангельмана
Риск повторного рождения детей с синдромом Ангельмана
Тип генетической передачи определяет тяжесть клинических проявлений: при делеции делеция в локусе 15 q11—q13 (1 вариант) более грубые умственные и двигательные нарушения, резистентные к терапии эпилептические приступы.
При этом виде мутации происходит снижение ГАМК – рецепторов типа А.
Критерии диагноза синдрома Ангельмана
Симптомы, полезные в качестве поддерживающих критериев, но отклонение от них не исключает диагноза СА:
Нормальное течение перинатального периода; нормальная окружность головы при рождении; отсутствие выраженных пороков развития.
Обязательные симптомы – встречаются в 100 % случаев при СА:
Часто встречающиеся симптомы (у 80 %)
Дополнительные критерии – у 20-80 %
Клинические проявления синдрома Ангельмана
Особенный черепно-лицевой и скелетный дисморфизм:
Часты соматические и вегетативные расстройства : склонность к запорам, пищеводный рефлюкс, диффузная гипотония мышц, плохая переносимость жары.
При проведении неврологического осмотра выявляются:
Косоглазие, диффузно сниженный мышечный тонус, повышение сухожильных рефлексов с конечностей, атаксия. Задержка моторного и психо – речевого развития умеренной и тяжелой степени тяжести.
Специфическая атаксическая походка: как марионетка – кукла на канатиках; «дерганная».
Резкие движения руками помогают удерживать тело в пространстве, балансировать.
Стереотипии – характерны необычные для здоровых повторяющиеся движения в виде взмахов рук, хлопанье в ладоши, кручения кистями.
Неэпилептический миоклонус – подергивания (вздрагивания) в конечностях.
Легкий тремор в покое.
Как распознать синдром Ангельмана
Частые симптомы синдрома Ангельмана :
Центральная нервная система
На МРТ и КТ картина неспецифическая:
Кожа
Редкие симптомы
Течение и прогноз при Синдроме Ангельмана
Лечение Синдрома Ангельмана
Медикаментозное лечение синдрома Ангельмана состоит:
Как общество относится к инвалидам, для чего необходимо адаптивное физическое развитие читайте в статье Паралимпиада в Сочи 2014.
Эпилептические приступы часто резистентны к терапии. С возрастом к 10 годам происходит урежение приступов.
В лечении эпилепсии при Синдроме Ангельмана используют:
Не используют в терапии СА в монотерапии из-за возможной аггравации приступов: фенитоин, карбамазепин, окскарбазепин, вигабатрин.
Но хороший эффект в комбинации
8. При фокальных моторных приступах – комбинация Вальпроата и Тегретола (15 – 25 мг/кг/сут).
9. При кортикальном миоклонусе эффект показал пирацетам в высоких дозах 140 мг/кг/сут внутривенно капельно.
10. Кетогенная диета.
11. Препараты ноотропного действия (осторожно).
12. Седативные препараты для улучшения сна, с подбором доз.
Приведем клинический пример, послуживший поводом этой публикации.
Родители дали согласие на публикацию этого примера.
Также на сайте читайте статью о наследственном Синдроме Рубинштейна – Тейби.
В нашем медицинском центре наблюдалось несколько пациентов с синдромом Ангельмана. Обращались с первичными жалобами на эпилептические приступы. После проведенных обследований выставлялся диагноз Синдром Ангельмана.
На приём в очередной раз обратились за выпиской для МСЭ родители с ребенком 6 лет 7 месяцев.
Жалобы при обращении: Последний приступ был на фоне течения ОРВИ с подъёмом температуры до 38,5 градусов С, более года назад в 22 часа, во время бодрствования, сидел на руках у мамы: вытянулся, заведение глаз в сторону, клонико – тонические судороги, длительность 25 минут, купировался после внутримышечного введения по Скорой помощи противосудорожного препарата. Был госпитализирован в инфекционный стационар.
Ранее генерализованный тонико – клонический приступ был 2 года 10 месяцев назад (02.2011 года).
Сохраняются миоклонические судороги в момент засыпания, почти каждый вечер, от 0 до 10 раз подряд, стали менее интенсивные. Иногда мама чувствует миоклонии только своей рукой, положив руку на тело ребенка.
Сохраняется задержка психоречевого и моторного развития, отмечается легкая положительная динамика.
Самостоятельно ходит с 4 лет. Шаткая походка. Двигательная активность улучшилась. Танцует под музыку. Пытается сам есть ложкой.
Стал больше понимать, выполняет простые команды, стал хаотично возить ручкой по листу. Книжки листает, непродолжительно занимается с игрушками, смотрит мультфильмы.
Периодически раздражителен. Беспокойный сон (двигательное беспокойство во время сна, поверхностный).
В терапии получает депакин – хроносферу по 250 мг на ночь (концентрация депакина в крови – 53 мкг/мл), суксилеп по 250 мг * 2 раза в день (концентрация суксилепа – 77 мкг/мл).
Анамнез. Беременность на фоне хронической гипоксии плода. Роды 2, в 37 недель, экстренное кесарево сечение. Масса при рождении 3400, длина 54 см, по Апгар 7/8 баллов.
Формула развития: голову удерживает с 2 месяцев, сидит самостоятельно с 12 месяцев, ходит у опоры с 2,5 лет. В речи к 6,5 годам – говорит 3-5 слов с 3,6 лет.
С рождения часто срыгивал, расценивали как течение дисбактериоза.
НСГ – признаки гидроцефалии.
В возрасте от 1 до 4 месяцев отмечались пароксизмы вскидывания рук перед собой, и как бы покачивание в руках. После курса Кортексина и Церебролизина, Цераксона все купировалось.
Диагноз ДЦП выставлен в 1 год 6 месяцев. Проводили курсы реабилитации, включая медикаментозное лечение прозерином, кортексином; физиолечение (электрофорез).
В 02.2012 году впервые возник приступ миоклоний с переходом во вторично – генерализованный приступ (возможно, это был миоклонический статус). На ЭЭГ – регистрировалась продолженная эпиактивность билатерально, синхронно. Задержка формирования корковых ритмов.
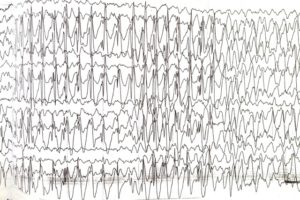 Эпиактивность при Синдроме Ангельмана
Эпиактивность при Синдроме Ангельмана
В 02.2012 году отмечался однократный эпизод, когда на фоне полного здоровья, после активного курса реабилитации, возникло состояние: активные миоклонии в плечевом поясе, кластерами; через 1 час появились падения назад с обмяканием, которые возникали после миоклоний. Между пароксизмами приходил в себя, то есть утрата контакта была при обмяканиях на несколько секунд. Пароксизмы продолжались по несколько секунд и длились до 1,5 суток, пока не ввели конвулекс. На фоне терапии конвулекса (35 мг/кг/сут) и дексазона пароксизмы стали уменьшаться. Приступы купированы через 3 суток.
В стационаре назначен дексаметазон №9, конвулекс по 75 мг * 2 раза в день длительно. Приступы были купированы, но отмечались побочные действия в виде интенсивного тремора.
ЭЭГ (с положительной динамикой) – регистрировалась эпиактивность по задним отведениям с короткими эпизодами билатеральной синхронизации.
С 05.2012 года конвулекс самостоятельно отменен. Приступы не отмечались.
Возобновились вздрагивания при засыпании с 07.2012 года.
На МРТ головного мозга (02.2010 года) – арахноидальная киста средней черепной ямки справа, внутренняя гидроцефалия.
МРТ головного мозга (02.2012 года) – расширение субарахноидальных щелей, киста правой височной доли (или недоразвитие).
Генетик (Москва): анализ на наследственные болезни обмена – без патологии.
Анализ на синдром Ангельмана сдан. Диагноз подтвержден в 06.2013 году.
 Генетический анализ на Синдром Ангельмана
Генетический анализ на Синдром Ангельмана
Заключение генетика: ish del (15)(q 11.2 q 11.2)(D15S11-, SNRPN-, D15S10-, GABR3-)
Отсутствуют 4 локуса хромосомы 15 в критической области синдромов Прадера – Вилли и Ангельмана (D15S11, SNRPN, D15S10, GABR3).
Переехал на постоянное место жительства из другого региона в 2012 году.
В ПЭЦ (противоэпилептическом центре) в 12.2012 году на ЭЭГ (фон + сон) – диффузные изменения биоэлектрической активности головного мозга. Мультирегиональная эпиактивность в височно – теменно – затылочных отделах больше справа и диффузная эпиактивность. Физиологические элементы сна – слабо выражены.
В лечении: подбор депакина – хроносферы.
Состояние с положительной динамикой. Миоклонии стали реже, улучшилась походка, стал активнее, говорит несколько слогов.
В ПЭЦ 06.2013 года на ЭЭГ (фон + сон) – региональная эпиактивность в левых лобно-теменных отведениях.
С 06.2013 года был введен суксилеп. Динамика положительная.
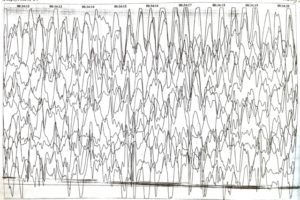 ЭЭГ синдром Ангельмана
ЭЭГ синдром Ангельмана
ЭЭГ (фон + сон) от 10.2013 года – региональная эпиактивность по затылочным отведениям; во сне регистрируется дельта активность по лобным отведениям с диффузным распространением.
 ЭЭГ синдром Ангельмана
ЭЭГ синдром Ангельмана
ЭЭГ (фон + сон) от 03.2014 года – диффузные изменения биоэлектрической активности головного мозга. В дремотном состоянии эпиактивности нет.
Травм, операций не было. Перенесенные заболевания – ОРВИ, ОКИ, ветряная оспа. Аллергия на сиропы, травы.
На осмотре невролога
Кожа и видимые слизистые чистые, бледные, волосы белесые.
Зев спокоен. Лимфоузлы не увеличены, отеков нет. В легких дыхание везикулярное, хрипов нет. Тоны сердца ясные ритмичные. ЧДД 20 в мин. ЧСС 84 уд в мин. Живот мягкий, безболезненный. Печень, селезенка не увеличены. Мочеиспускание свободное. Стул оформленный.
Обращает на себя внимание фенотип : маленький череп, гипоплазия средней части лица, макростомия (широкий рот), широкие межзубные промежутки, атаксия, задержка психо –речевого развития, мышечная гипотония, приступы смеха, сухожильная гиперрефлексия.
Неврологический статус
Состояние стабильное. Сознание ясное, реакция на осмотр не адекватная. Улыбчив, добродушен, легко идёт на контакт. Команды выполняет избирательно, не все понимает. Гиперактивен.
Череп микроцефальной формы (микробрахиоцефалия), окружность головы 45 см.
Глазные щели симметричны, объем движения глазных яблок полный, реакция зрачков на свет живая D=S. Лицо симметрично, носогубные складки не сглажены. Слух не нарушен. Глотает самостоятельно, не поперхивается. Язык по средней линии, высунут изо рта.
Расстройства чувствительности нет.
Координационные пробы не понимает. Походка атаксическая, ходит на широко расставленных ногах.
Менингеальных знаков нет.
Диагноз : Миоклонический статус при непрогрессирующей энцефалопатии на фоне хромосомной патологии, синдрома Ангельмана. Органическое поражение ЦНС, атонически – астатический синдром, грубая задержка психоречевого развития, GMFCS IV уровень.
Итак, в приведенном примере мы увидели все характерные черты синдрома Ангельмана; проследили, как начинались и менялись эпилептические приступы и картина ЭЭГ; эффективность терапии. Встретив пациента с похожими симптомами, мы заподозрим и направим к генетикам на уточнения диагноза, выберем более верную тактику лечения.
На видео наш пациент ребенок с Синдромом Ангельмана
Научная электронная библиотека

Юров И. Ю., Ворсанова С. Г., Воинова В. Ю., Чурносов М. И., Юров Ю. Б.,
1.4.2.1. Микроделеционные и микродупликационные синдромы
Ранее, при исследовании кариотипа классическими цитогенетическими методами, считалось, что структурные аномалии хромосом (инсерции, инверсии, делеции, дупликации, транслокации) встречаются значительно реже, чем численные. Выявление структурных аномалий хромосом зависит от их размера, локализации, типа перестройки (регулярная или мозаичная), а также метода, используемого для исследования. При этом разрешающая способность классических цитогенетических методов составляет от 5–7 млн пн. С внедрением современных молекулярно-цитогенетических технологий исследования хромосом (генома) стали возможны с разрешающей способностью от 1000 пн и меньше, что позволяет обнаружить субмикроскопические перестройки и уточнить координаты геномных нарушений, в том числе микроделеций и микродупликаций, и число обнаруженных структурных перестроек, значительно возросло. Это, в свою очередь, позволяет получать информацию о генах, находящихся в исследуемых участках, и в дальнейшем проводить биоинформатический анализ с целью определения генов-кандидатов патологических фенотипических проявлений у детей.
Данные аномалии часто выявляются при анализе генома детей с недифференцированными формами умственной отсталости, микроаномалиями развития (МАР) и врождёнными пороками развития (ВПР). Известно, что структурные аномалии хромосом ассоциированы с определёнными фенотипическими проявлениями, в том числе, как и с наиболее частыми микроделеционными/микродупликационными синдромами, так и с редкими микроаномалиями. Несмотря на это, даже при наиболее часто встречающихся микроделеционных/микродупликационных синдромах, затрагивающих области, в которых локализованы более 100 генов, вклад отдельных генов в формирование патологических фенотипических проявлений ещё предстоит изучать. Наиболее часто встречающиеся микроделеционные/микродупликационные синдромы и аномалии представлены в таблице 2. У многих синдромов, описанных с внедрением современных молекулярно-цитогенетических технологий, частота не известна. Ниже даётся описание отдельных микроделеционных/микродупликационных синдромов и аномалий.
Синдром микроделеции 1p36
При данном синдроме наблюдаются задержка моторного развития, расстройства аутистического спектра; некоторые исследователи отмечают, что 25 % пациентов могут ходить самостоятельно, широкой походкой, примерно к 2–7 годам. Экспрессивная речь отсутствует в 75 % случаев, понимание обращенной речи ограничено определенными ситуациями. Стремление к коммуникации в ранние периоды развития проявляется слабо, но улучшается со временем при расширении используемых жестов. Многие исследователи выявляют следующие клинические признаки: резкие перемены настроения, самоповреждающее поведение, стереотипии и МАР: прямые брови, микроцефалия, широкая переносица, низко расположенные аномальной формы ушные раковины, клинодактилия, небольшие ступни, а также отмечают пороки сердечно-сосудистой системы (ССС).
При микроделеции 1q41q42 наблюдаются умеренная умственная отсталость, аутизм, судороги, микроцефалия, косолапость, МАР: гипотелоризм глазных щелей, вывернутые вперёд ноздри.
Синдром микроделеции 2q37
Описано более 100 клинических случаев с микроделецией участка 2q37. У этих индивидуумов обычно отмечается лёгкая или умеренная задержка психического развития, у 30 % пациентов имеются черты аутизма. Дети обычно невысокого роста с гипермобильными суставами, сколиозом, синдактилией кистей и/или стоп. Пороки сердца встречаются у 35 %, нередко выявляются пороки почек. К характерным лицевым аномалиям при синдроме микроделеция 2q37 относят следующие: брахицефалия, круглое широкое лицо, редкие волосы на голове, широкие лоб и нос с расщеплением кончика, глубоко посаженные глаза, редкие высокие арочные брови, готическое нёбо; отмечают также укороченные фаланги пальцев рук и ног, экзему.
При данной микроделеции наблюдается умственная отсталость средней тяжести, отмечаются аутистические черты, МАР: микроцефалия, длинное узкое лицо, большие ушные раковины, аномально сформированная спинка носа, а также атаксия, пороки сердца и почек. Нередко отмечают аутистические проявления.
При данной микродупликации наблюдается умственная отсталость средней тяжести, встречаются аутистические проявления, задержка роста, задержки психомоторного (ЗПМР) и психоречевого развития (ЗПРР), микроцефалия, лицевые аномалии: высокий лоб, низко расположенные ушные раковины, короткая шея, аномалии глазных щелей, а также гипотония с мышечной гипотрофией, пороки ССС.
Синдром Вольфа-Хиршхорна (делеция в участке 4р16)
При данной микроделеции наблюдаются умственная отсталость различной степени тяжести, задержка роста, ЗПМР и ЗПРР, микроцефалия, лицевые микроаномалии: высокий лоб, эпикант, маленький рот с опущенными углами, клювовидный нос с выступающим надпереносьем, микрогения, низко расположенные деформированные ушные раковины с преарикулярными складками, короткая шея, а также пороки ССС, почек, желудочно-кишечного тракта, гипоспадия, гипотония с мышечной гипотрофией, судороги. Иногда встречаются пороки мозга в виде агенезии или гипоплазии мозолистого тела, гипоплазии мозжечка. Характерным признаком синдрома является воронкообразное углубление в области крестца (sinus sacralis). Частота этого синдрома в популяции 1:90000 – 100000 по данным публикаций различных авторов. Встречаются аутистические расстройства.
Синдром «крика кошки» («cri du chat», делеция в участке 5p15.2)
Наибольшее отставание в развитии при этом синдроме наблюдается при навыках, которые требуют мобильности, ловкости и вербальной коммуникации. По сравнению с мелкой моторикой, крупная моторика рук относительно сохранна и дети способны совершать «машущие» движения рукой или поймать мяч. Отсутствие речи компенсируется у 2/3 детей при помощи невербальных методов коммуникации, примерно 50 % детей способны использовать язык жестов для сообщения основных потребностей. Основным диагностическим признаком синдрома является «крик кошки», связанный с изменением гортани (сужение, мягкость хрящей, отечность или складчатость слизистой, уменьшение надгортанника). Наблюдаются также следующие клинические признаки: умственная отсталость, микроцефалия, низко расположенные деформированные ушные раковины, микрогения, гипертелоризм и антимонголоидный разрез глазных щелей, эпикант, косоглазие, гипотония мышц, пороки ССС, иногда выявляют аринэнцефалию, гипоплазию мозжечка, микрогирию больших полушарий, пороки развития почек и желудочно-кишечного тракта. Частота синдрома в популяции 1:45000.
Синдромы Вольфа-Хиршхорна и «крика кошки» относятся к делеционным синдромам, но довольно часто геномные микроаномалии затрагивают только критические сегменты хромосом (4р16 и 5р15, соответственно), и тогда можно их отнести к микроделеционным синдромам.
При данной микродупликации наблюдаются умственная отсталость различной степени тяжести, аутизм, черепно-лицевые аномалии: макроцефалия, гипертелоризм глазных щелей, аномалии ушных раковин.
Синдром Вильямса (делеция в участке 7q11.23)
Основными проявлениями синдрома являются хрипловатый голос, отсутствие чувства дистанции при общении. У пациентов с синдромом Вильямса наблюдается слабая зрительно-моторная интеграция, в результате чего вместо целостной картинки они видят ее отдельные составные части. Кроме того, у больных выявлены макроцефалия, макрокрания, мышечная гипотония, МАР: широкий лоб, глубоко посаженные глазные щели, широкая короткая переносица, оттопыренные ушные раковины, колобома, аномалии зубов; потеря слуха, тревожность, дефицит внимания с гиперактивностью (СДВГ), пороки ССС. Многие дети с этим синдромом могут играть на музыкальных инструментах, общительны, не имеют задержки в речевом развитии. Частота 1:10000 – 20000 по данным публикаций различных авторов.
При данной микроделеции наблюдаются ЗПРР, расстройства аутистического спектра, МАР: синофриз, широкий нос, ретрогнатия, широкие пальцы с короткими дистальными фалангами.
Синдром микродупликации 15q11q13
Следует отметить, что микродупликацию 15q11q13 можно отнести к часто встречающимся цитогенетическим аномалиям при аутизме. По данным литературы, указанная микроперестройка встречается примерно у 1 % детей с аутизмом, и она индексирована в базе данных OMIM (Online Mendelian Inheritance in Man) (OMIM:608636) как генетически обусловленное состояние. Имеются исследования, направленные на приоритизацию генов-кандидатов психических нарушений у пациентов с перестройками в этом участке, включая и микродупликацию.
Синдром Ангельмана (делеция или унипарентальная дисомия в участке 15q11.2q13 материнского происхождения)
Нарушения в данном участке связаны с таким генетическим заболеванием, как синдром Ангельмана при нарушении в хромосоме материнского происхождения. При перестройке материнского происхождения у детей часто наблюдаются эпилепсия, атаксия, умственная отсталость и характерные лицевые микроаномалии. Основной значимой характеристикой синдрома Ангельмана является чрезмерно положительное настроение с постоянной улыбкой и смехом у ребёнка. Выявляются фокальные стереотипии, однако не наблюдаются специфичные, часто повторяющиеся стереотипные движения. Дети заинтересованы в социальных взаимодействиях, многие пациенты стремятся к коммуникации, несмотря на выраженные нарушения речи. Пациенты испытывают трудности во взаимодействии по причине слабого понимания социальных и эмоциональных сигналов. Психические нарушения зависят от происхождения перестройки, а именно, от того, является ли делетированный участок материнским или отцовским. Это позволяет говорить о том, что эпигенетический феномен геномного импринтинга играет значимую роль в этиологии и патогенезе психических нарушений.
Синдром Прадера-Вилли (делеция или унипарентальная дисомия в участке 15q11.2q13 отцовского происхождения)
Нарушения поведения при данном синдроме проявляются в резких перепадах настроения, упорстве, соответствующем поведении, обсессивно-компульсивных характеристиках, а также сложностью в отвлечении от ежедневно повторяемых рутинных событий. Синдром Прадера-Вилли ассоциирован с повышенным риском психических нарушений. Пациенты с синдром Прадера-Вилли при отсутствии отцовской копии указанного участка хромосомы 15 (унипарентальная дисомия) подвержены различным психическим расстройствам.
При данной микроделеции наблюдаются умственная отсталость, низкорослость, микроцефалия, пониженный мышечный тонус (гипотония), скелетные аномалии, гипоспадия.
Синдром микроделеции 16p11.2
Перестройки в участке 16p11.2, по данным литературы, ассоциированы с аутизмом и лёгкой формой умственной отсталости. При исследовании детей с расстройствами аутистического спектра микроделеция участка 16p11.2 выявляется в 0,4–1,2 % случаев. Чаще обнаруживают эту микроделецию de novo; тем не менее, микроделеция также может передаваться от родителей к ребёнку. Микроделеции участка 16p11.2 встречаются чаще при аутизме по сравнению с микродупликациями этого же участка. У пациентов с микроделецией в данном участке отмечают более тяжёлые фенотипические проявления по сравнению с микродупликацией. Выявляются следующие врождённые проявления: задержка речевого развития (ЗРР), отставание в физическом развитии, двигательные нарушения, эпилепсия, пороки сердца, ожирение. Среди МАР отмечают низко расположенные ушные раковины и частично перепончатые пальцы.
Синдром Смит-Маженис (делеция в участке 17p11.2)
Для микроделеции участка 17p11.2 характерны различные фенотипические проявления. Эти признаки были объединены в синдром Смит-Маженис, индексированным в OMIM (182290). Для синдрома характерны черепно-лицевые аномалии (широкое/плоское лицо, широкая переносица, выпуклый лоб, сросшиеся брови, низко расположенные ушные раковины), широкие короткие кисти, плоскостопие, брахидактилия, а также нарушения развития и поведения. Черты аутизма присутствуют более чем у 50 % детей с данным синдромом. У 80 % пациентов проявляется самоповреждающее поведение (аутоагрессия), включая онихотилломанию, кусание запястий, качание головой, а также повышенную толерантность к боли, нарушение сна. Навыки импрессивной речи, как правило, выше, чем экспрессивной. Хриплый голос может являться диагностическим маркером синдрома. Использование языка жестов значительно способствует улучшению коммуникативных способностей ребенка. Выявляют умственную отсталость от умеренной до тяжёлой степени.
Следует сказать и про микродупликацию в этом участке, которая ассоциирована с синдромом Потоцки-Лупски, индексированным в OMIM (610883). Заболевание проявляется гипотонией мышц, врождёнными пороками развития, лицевыми аномалиями, пороками ССС, низким ростом и различными нервными и психическими нарушениями. По данным исследований, аутизм встречается у этих детей в 65 % случаев.
Синдром микроделеции 22q11.2
Синдром микроделеции участка 22q11.2 ассоциируется с аутизмом, велокардиофациальным синдромом и синдромом ДиДжорджи. Частота его составляет 1:4000 новорождённых. У детей с такой перестройкой выявляют нарушение психического и моторного развития, отмечают медленный рост и гипотонию. К менее распространённым нарушениям при нём можно отнести аномалии ССС, расщелину нёба, лицевые МАР, нарушение иммунитета. Аутистические проявления наблюдаются также у 15–25 % пациентов с микродупликацией в данном участке.
Синдром микроделеции 22q13.3
Синдром микроделеции 22q13.3 известен также как синдром Фелан-МакДермид. Он с одинаковой частотой встречается у лиц мужского и женского пола. К характерным признакам данного синдрома можно отнести неонатальную гипотонию, микроаномалии развития, задержку или полное отсутствие речи, аутизм и эпилепсию. К основным фенотипическим проявлениям данного синдрома относятся следующие: большие ушные раковины, диспластичные ногти, широкий лоб, острый подбородок. При исследовании нескольких групп детей с микроделецией в участке 22q13.3 аутистические черты были выявлены у 85 % пациентов.
Основные характерные особенности, свойственные известным микроделеционным и микродупликационным синдромам, представлены в таблице 2.
Характерные особенности некоторых часто встречающихся микроделеционных* и микродупликационных* синдромов и аномалий



