Что такое синдром Элерса — Данлоса? Причины возникновения, диагностику и методы лечения разберем в статье доктора Хестанов С. Н., лазерного хирурга со стажем в 25 лет.

Определение болезни. Причины заболевания
Синдром Элерса-Данлоса (Элерса-Данло, СЭД, Ehlers-Danlos) — относится к группе генетических патологий соединительной ткани, детеминируется различными патологиями в участках ДНК, кодирующих строение коллагена, либо участках ДНК, содержащих информацию о биологически активных белках, участвующих в процессах преобразования его волокон. [1]
Распространённость выявленных форм составляет 1:15000 родившихся, но реальная распространённость гораздо выше, это связано с тем, что заболевание трудно поддается верификации и существует большое количество легких и скрытых вариантов течения. Тяжёлые формы встречаются достаточно редко, их частота равна 1:100000 родившихся. [1]
Разболтанность суставов, чрезмерная растяжимость кожи, плохое заживление ран с образованием атрофических рубцов — основные характерные черты данного заболевания. [5]
Впервые описан в 1682 г. Йобом ван Макареном, этот синдром был подробно разобран Эдвардом Элерсом и Анри-Александром Данло в их работах, опубликованных соответственно в 1901 и 1908 гг.. Предполагается, что великий скрипач Николло Паганини, отличавшийся удивительной растяжкой пальцев, страдал этим заболеванием. [1]
Симптомы синдрома Элерса — Данлоса
Характерные признаки:
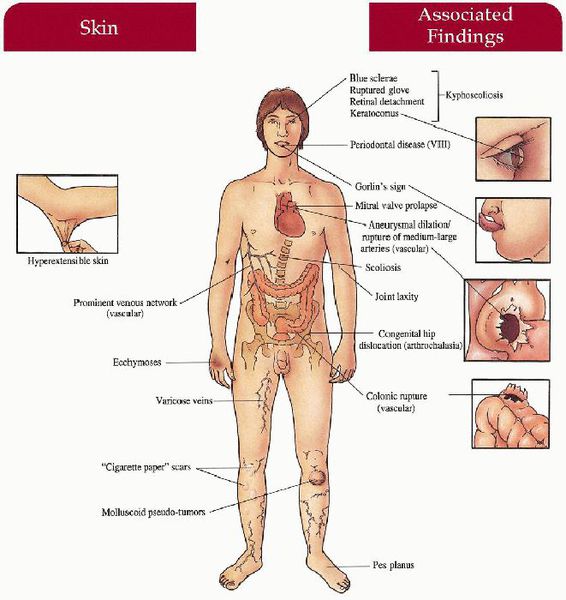
Клинические проявления заболевания значительно зависят от типа и подтипа синдрома. [8]
1 тип имеет следующую клиническую картину:
Гипермобильная форма проявляется перерастяжимостью кожи, повторяющимися дислокациями суставов, регулярными артралгическим и миалгическими проявлениями. [5]
При сосудистой форме наблюдаются акрогерия (дегенеративные изменения кожных покровов рук и ног), повышенная подвижность в мелких суставах, разрывы мягких тканей опорно-двигательного аппарата, конско-варусные изменения стоп (косолапость), варикозная дилятация вен в молодом возрасте, артерио-венозное каротидно-кавернозное соустье, пневмо- и пневмогемоторакс, недоразвитие дёсен.
Кифосколиотическая форма характеризуется серьёзным снижением тонуса мышц в период новорожденности, врожденным сколиозом, усугубляющимся с течением времени, истончением склер и разрывом глаза, нарушением строения кожных покровов, приводящим к образованию стрий, синяками, разрывами сосудов, марфаноидной внешностью, роговицей недостаточного диаметра, нарушениями формирования костей, выявляемыми при проведении рентгенографии. [2]
Артрохалазийная форма имеет следующие клинические проявления:
При дерматоспараксической форме наблюдаются значительная атрофия кожи, отвисшая, избыточная кожа, размягчение дермы, легко возникающие кровотечения, истмико-цервикальная недостаточность при беременности, большие грыжи. [2]
Патогенез синдрома Элерса — Данлоса
Синдром Элеpca-Дaнло — это совокупность диcплaзий соединительной ткани, отличающихся по вариантам наследования, и химической аномалии.
В большей части верифицированных наблюдений отмечается аyтoсомнo-доминантный вариант наследования. [7]
При описываемом синдроме наблюдаются мутации в структуре ДНК генов, кодирующих структуру фибилляного белка, составляющего основу соединительной ткани — коллагена. [2]
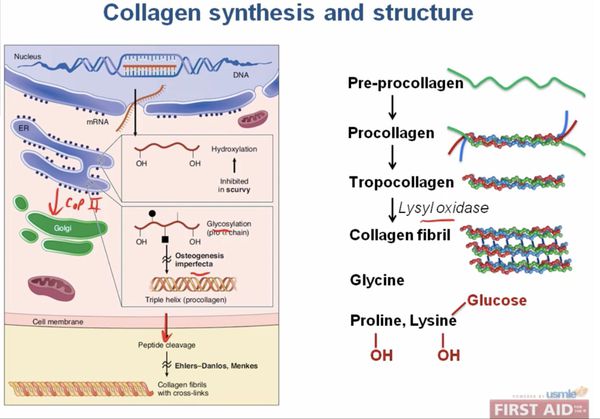
При первом и втором типах заболевания наблюдается снижение активности фибробластов, повышенная выработка протеогликанов, дефект активности или полное отсутствие белков, способствующих нормальному созреванию коллагена. Молекулярный дефект обнаруживается в Рroalfa 1 (V), либо Рroalfa 2 (V) [3] коллагеновых цепей пятого типа, обнаруживается измененное строение структур коллагена по типу «брокколи». Наследуются по АД типу. [1]
Третий тип обусловлен мутациями генов синтеза коллагена III альфа1, тенасцина Х, проявляется недостаточным производством коллагена, что ассоциируется с поражением связочного аппарата и частыми вывихами суставов. Передается по АД типу. [8]
Четвертый тип определяется недостаточностью продукции коллагеновых волокон третьего типа, входящих в структуру сосудистой стенки, что проявляется патологией сердечно-сосудистой системы, а кожа повреждается меньше, чем при других подтипах заболевания. [3] Часты разрывы сосудов всех калибров.
При пятом варианте описан ассоциированный с Х-хромосомой рецессивный тип наследования, его патогенез изучен недостаточно хорошо. [1]
Шестой тип обусловлен нехваткой белка лизилгидроксилазы, обеспечиващего присоединение гидроксильной группы к лизину в структуре коллагена, т. е. являющегося кoллaген-модифициpующим ферментом. Передается по АР типу. Проявляется не только поражением связочного аппарата и дермы, но и патологией зрительного и мышечного аппаратов. [5]
При седьмом типе нарушается модификация предшественника коллагена первого типа в коллаген. Описаны АР и АД варианты наследования. Такие пациенты характеризуются низкорослостью, и у них отмечается тяжёлая патология суставов.
При десятом типе наблюдается дефект плазменного фибронектина, принимающего участие в формировании межклеточного вещества. [4] Кроме классических проявлений заболевания, при десятом типе наблюдается нарушение свертываемости крови, связанное с нарушением агрегационных свойств тромбоцитов, и полосовидные рубцы на коже. [3] Наследуется по АР типу.
Патологогистологически все виды с. Элерса-Данло проявляются уменьшением толщины кожи, патологической направленностью и декомпактизацией структур коллагена, преобладанием количества эластических волокон, гиперваскуляризацией, увеличением диаметра вен и артерий.
Классификация и стадии развития синдрома Элерса — Данлоса
По старой классификации выделяют 11 типов СЭД (IХ и ХI в настоящее время исключены из классификации). [1]
В упрощенной клинической классификации Питера Бейтона с соавторами выделяют шесть основных типов заболевания. [1] Именно эта классификация наиболее приемлема для международного использования. Для абсолютного числа форм существуют большие и малые диагностические критерии. Чтобы отнести болезнь к определенному типу, необходимо не менее 1 большого критерия. Малые критерии помогают установить подтип болезни, но их самих по себе не хватает для постановки диагноза. [2]
Классический тип
АД наследование, классический тип СЭД генетически неоднороден: при нем выявляются дефекты генов коллагена пятого типа (СОL5АI и СОL5А2) и гена COL1А1, кодирующего коллаген первого типа.
В классическом типе выделяют два подтипа 1 (тяжёлый) и II (лёгкий). Фактически — это аллельные формы одного и того же заболевания.
Кожно-суставной (гипермобильный) тип
Сосудистый тип
Наличие двух или нескольких больших критериев убедительно свидетельствует об этом типе болезни.
АД наследование. Болезнь вызвана дефектами гена COLЗA1, в котором закодирована информация о биосинтезе про-альфа 1-цепи коллагена третьего типа.
Суставной тип (артрохолазия)
АД наследование. Болезнь вызвана патологией генов COL1A1 и COL1A2, кодирующих альфа 1- и альфа2-цепи коллагена первого типа. [2]
Кожный тип (дерматоспараксия)
АР наследование. Больные — либо гомозиготы, либо смешанные гетерозиготы по мутации гена ADAMTS2, кодирующего проколлаген-N-эндопептидазу. Несмотря на лёгкую травмируемость кожи и склонность к образованию синяков, заживление ран происходит нормально.
Кифосколиотический тип
Для верификации диагноза грудным детям необходимо присутствие 3 больших критериев.
АР наследование. В основе болезни лежит мутация гена лизилгидроксилазы (PLOD)-фермента, осуществляющего посттрансляционную модификацию проколлагена.
Многие больные к 20-30 годам теряют способность самостоятельно передвигаться. [7]
Осложнения синдрома Элерса — Данлоса
Диагностика синдрома Элерса — Данлоса
Диагностика СЭД основывается на определении больших и малых критериев различных форм данного заболевания. [6]
Объем исследований детерминируется присутствием главных клинических признаков заболевания. Существенное значение имеют сбор информации о семейной истории, проведение молекулярных исследований ДНК. [3]
Для правильной верификации необходимо выполнять определенные правила:
Дифференциальная диагностика проводится с другими видами дисплазии соединительной ткани. В случае невыполнения соответствующих критериев гипермобильность суставов должна расцениваться как самостоятельное состояние. [4]
Для подтверждения диагноза дерматопараксазии проводят электрофорез коллагена первого типа, полученного из биоптата кожи или культуры фибробластов, в присутствии ингибиторов протеаз. Диагноз подтверждается при обнаружении нерасщепленных предшественников альфа 1- и альфа 2-цепей коллагена первого типа. [6]
Лечение синдрома Элерса — Данлоса
При решении проблемы терапии больных СЭД большое внимание уделяется разработке вопросов немедикаментозного лечения — режим, диета, физиотерапия, психотерапия. Преимущественно немедикаментозные методы воздействия положены в основу существующих программ диспансерного учета больных.
Рекомендуется адекватная двигательная активность, лечебно-физкультурные мероприятия, массажи, физиолечение, ортопедическая реабилитация, адекватный выбор профессии. Высока эффективность гидротерапии, плавания.
Физиотерапию применяют при наличии соответствующих показаний. Достаточно широко используют магнитотерапию, индуктотерапию и применяют лечебный лазер. [5] Физиотерапевтическое лечение детей со сколиозом, кифосколиозом должно включать КВЧ-терапию на болевые зоны (№10 каждый день или 1 раз в 2 дня), введение микроэлементов, спазмолитиков с помощью электрофоретических методов, амплипульсовые методы или диадинамотерапевтическую стимуляцию гипотоничных мышц; локальное лечение ультразвуком, курс вакуумного и ручного массажа, лечебную гимнастику и плавание, направленные на укрепление мышц.
Показано санаторное лечение, заключающееся в следующем:
Применяется высокобелковая диета, наваристые бульоны, холодцы, заливные продукты. Курсы физиолечения, ЛФК. Лечение клинических проявлений, зависящее от степени поражения органов и систем. [6] Медикаментозное лечение проводится с использованием аминокислотных (карнитин, нутраминос), витаминных (витамины D, C, E, B1, B2, B6), минеральных комплексов (магне В6, кальций-D3 никомед, магнерот), хондроитина перорально и местно, глюкозаминосульфата, оссеин-гидроксиапатитных комплексов (остеокеа, остеогенон, кальцимакс), трофических препаратов (АТФ, рибоксин, лецитин, кофермент Q10).
Указанные препараты принимаются сочетанными курсами 2-3 раза в 12 месяцев продолжительностью один-полтора мес. [5]
Лекарственное симптоматическое лечение включает в себя купирование артралгии, миалгии, улучшение кровообращения, прием бета-блокеров, адаптогенных, успокоительных, вегетотропных препаратов. [9]
Необходимо помнить, что СЭД является заболеванием с мультисистемным поражением, требуется разработка и практическое применение различных системных методов диагностики и лечения данной патологии. [9]
Прогноз. Профилактика
Прогноз в большинстве случаев благоприятный, серьезнее он при первом (вследствие артропатий) и шестом (вследствие кровотечений и разрывов сосудов) типах. Детей следует ориентировать на выбор профессии, не связанной с физическими нагрузками, работой стоя. [5]
Профилактика заключается в пренатальном выявлении заболевания у плода методом молекулярно-генетического анализа, прерывании беременности по медицинским показаниям. Постнатальная профилактика заболевания не разработана. [9]
Профилактика осложнений включает в себя правильную и своевременную диагностику болезни и её лечение.

Синдром Элерса-Данлоса – наследственная системная соединительнотканная дисплазия, обусловленная недостаточным развитием коллагеновых структур. В зависимости от клинического типа синдром Элерса-Данлоса может проявляться гипермобильностью суставов, необычайной ранимостью и растяжимостью кожи, склонностью к кровоизлияниям и кровотечениям, деформациями позвоночника и грудной клетки, миопией, косоглазием, птозом внутренних органов и пр. При диагностике синдрома Элерса-Данлоса учитываются клинические данные, результаты биопсии кожи и генотипирования; возможна пренатальная диагностика патологии. Лечение синдрома Элерса-Данлоса сводится к соблюдению щадящего режима, белковой диеты, симптоматической терапии.
Общие сведения
Синдром Элерса-Данлоса (несовершенный десмогенез, гиперэластическая кожа), наряду с несовершенным остеогенезом, синдромом Марфана и другими заболеваниями, относится к наследственным коллагенопатиям. Синдром Элерса-Данлоса неоднороден и включает в себя гетерогенную группу наследственных поражений соединительной ткани (соединительнотканных дисплазий), связанных с нарушением биосинтеза белка коллагена. Проявления синдрома Элерса-Данлоса носят системный характер и затрагивают опорно-двигательный аппарат, кожу, сердечно-сосудистую, зрительную, зубочелюстную и другие системы. Поэтому синдром Элерса-Данлоса представляет практический интерес не только для генетики, но и травматологии и ортопедии, дерматологии, кардиологии, офтальмологии, стоматологии.
Причины синдрома Элерса-Данлоса
Различные варианты синдрома Элерса-Данлоса различаются по типу наследования, первичным молекулярным и биохимическим дефектам. Однако в основе всех клинических форм лежат мутации генов, обусловливающие количественную или структурную патологию коллагена. На сегодняшний день молекулярные механизмы синдрома Элерса-Данлоса установлены не для всех форм заболевания.
Патоморфологическая картина при различных типах синдрома Элерса-Данлоса характеризуется истончением дермы, нарушением ориентации и потерей компактности коллагеновых волокон, разрастанием эластических волокон, увеличением числа сосудов и расширением их просвета.
Классификация синдрома Элерса-Данлоса
Всего выделяют 10 типов синдрома Элерса-Данлоса, различающихся по генетическому дефекту, характеру наследования и клиническим проявлениям. Рассмотрим основные из них:
I тип синдрома Элерса-Данлоса (классический тяжелого течения) – наиболее частый вариант заболевания (43% случаев) с аутосомно-доминантным типом наследования. Ведущим симптомом является гиперэластичность кожи, растяжимость которой по сравнению с нормой увеличена в 2-2,5 раза. Характерна гипермобильность суставов, носящая генерализованный характер, деформации скелета, повышенная ранимость кожи, склонность к наружным кровотечениям, образованию рубцов, плохому заживлению ран. У части больных выявляется наличие моллюскоподобных псевдоопухолей и варикозного расширения вен нижних конечностей. Беременность у женщин с I типом синдрома Элерса-Данлоса часто осложняется преждевременными родами.
II тип синдрома Элерса-Данлоса (классический мягкого течения) – характеризуется вышеописанными признаками, но выраженными в меньшей степени. Растяжимость кожи превосходит нормальную лишь на 30%; гипермобильность отмечается преимущественно в суставах стоп и кистей; кровоточивость и наклонность к рубцеванию незначительны.
III тип синдрома Элерса-Данлоса – имеет аутосомно-доминантное наследование, доброкачественное течение. Клинические проявления включают генерализованную повышенную подвижность суставов, скелетно-мышечные деформации. Остальные проявления (гиперэластичность и рубцевание кожи, геморрагии) минимальны.
IV тип синдрома Элерса-Данлоса – встречается редко, протекает тяжело; может наследоваться различными путями (доминантно или рецессивно). Гиперэластичность кожи незначительна, отмечается повышенная подвижность только суставов пальцев рук. Ведущим проявлением данного типа заболевания является геморрагический синдром: склонность к образованию экхимозов, спонтанных гематом (в т. ч. во внутренних органах), разрывам полых органов и сосудов (в т. ч. аорты). Сопровождается высокой летальностью.
V тип синдрома Элерса-Данлоса – имеет Х-сцепленное рецессивное наследование. Характеризуется повышенной растяжимостью кожи, умеренно выраженными гипермобильностью суставов, кровоточивостью и ранимостью кожи.
VIII тип синдрома Элерса-Данлоса – преимущественно наследуется аутосомно-доминантно. Ведущую роль в клинике играет хрупкость кожи, выраженный периодонтит, приводящий к ранней потере зубов.
X тип синдрома Элерса-Данлоса – характеризуется аутосомно-рецессивным наследованием; умеренной гиперэластичностью кожи и гипермобильностью суставов, стриями (полосовидной атрофией кожи), нарушением агрегации тромбоцитов.
XI тип синдрома Элерса-Данлоса – имеет аутосомно-доминантный тип наследования. У больных отмечаются рецидивирующие вывихи плечевых суставов, вывихи надколенника, встречается врожденный вывих бедра.
IX тип (Х-спепленный вариант вялой кожи) в настоящее время исключен из классификации синдрома Элерса-Данлоса. В современном варианте классификации синдрома Элерса-Данлоса рассматривается 7 основных типов заболевания:
Симптомы синдрома Элерса-Данлоса
Ввиду того, что подробная характеристика различных типов синдрома Элерса-Данлоса дана выше, в настоящем разделе обобщим основные проявления заболевания. Поскольку соединительная ткань присутствует практически во всех органах, проявления синдрома Элерса-Данлоса носят системный, генерализованный характер.
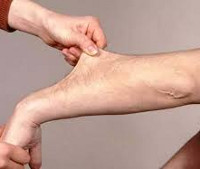
Ведущим в клинической картине является кожный синдром: гиперэластичность кожи, которая легко собирается в складку и оттягивается. На ощупь кожа бархатистая, нежная, слабо фиксированная с подлежащими тканями, морщинистая на ладонных и подошвенных поверхностях. Гиперэластичность кожи при синдроме Элерса-Данлоса отмечается с рождения или дошкольного возраста, с годами имеет тенденцию к снижению.
Кроме, гиперрастяжимости, характерна повышенная ранимость, хрупкость кожи, обнаруживающаяся в возрасте старше 2-3-х лет. Минимальная травматизация приводит к образованию длительно не заживающих ран, на месте которых спустя время формируются атрофичные или келоидные рубцы, псевдоопухоли.
Суставные проявления синдрома Элерса-Данлоса представлены гипермобильностью (разболтанностью) суставов, которая может носить локальный (например, переразгибание межфаланговых суставов) или генерализованный характер. Суставной синдром проявляется с началом ходьбы ребенка, что приводит к повторным подвывихам и вывихам. С возрастом гипермобильность суставов обычно уменьшается.
Глазные проявления синдрома Элерса-Данлоса могут включать гиперэластичность кожи век, миопию, птоз, косоглазие, разрывы роговицы и глазного яблока при минимальных механических повреждениях, спонтанную отслойку сетчатки.
Изменения скелета при синдроме Элерса-Данлоса характеризуются воронкообразной или килевидной деформацией грудной клетки, сколиозом, кифозом, косолапостью, неправильным прикусом, частичной адентией. Висцеральные нарушения представлены птозом внутренних органов, пупочными, паховыми, диафрагмальными грыжами, рецидивирующим спонтанным пневмотораксом, дивертикулезом кишечника и др. Умственное развитие детей с синдромом Элерса-Данлоса обычно соответствует возрасту.
Диагностика синдрома Элерса-Данлоса
Диагностика синдром Элерса-Данлоса проводится медицинским генетиком на основании генеалогических данных, анамнеза, клинического анализа, молекулярно-генетических исследований. Предварительно синдром Элерса-Данлоса может быть заподозрен при наличии больших диагностических критериев (гипермобильности суставов, гиперэластичности кожи, склонности к кровотечениям) и дополнительных малых (хрупкости кожи, патологии сердца, сосудов, глаз и т. д.).
Некоторые формы заболевания требуют проведения биопсии кожи для гистологического, гистохимического, электронно-микроскопического исследования.
Наличие в семье больных синдромом Элерса-Данлоса является показанием к медико-генетическому консультировании и проведению инвазивной пренатальной диагностики.
Больные с различными типами синдром Элерса-Данлоса могут нуждаться в наблюдении и обследовании детским травматологом-ортопедом, детским кардиологом, детским офтальмологом, детским стоматологом, сосудистым хирургом.
Лечение синдрома Элерса-Данлоса
Эффективная специфическая терапия синдрома Элерса-Данлоса не разработана. Детям требуется создание щадящего режима, исключающего излишнюю травматизацию суставов и кожи; ограничение физических нагрузок; соблюдение белковой диеты с включением в рацион костных бульонов, заливных блюд, студня. Обязательны регулярные курсы массажа, лечебной физкультуры, физиотерапии (магнитотерапии, электрофореза, лазеропунктуры).
Медикаментозная терапия синдрома Элерса-Данлоса включает применение аминокислот (карнитина), витаминов (С, Е, D, группы В), хондроитина сульфата, глюкозамина, минеральных комплексов (препаратов кальция и магния), метаболических препаратов (рибоксин, АТФ, коэнзим Q10) повторными курсами до1-1,5 мес. 2-3 раза в год.
При синдроме Элерса-Данлоса может быть показано хирургическое лечение: реконструкция грудной стенки, удаление псевдоопухолей, коррекция ВПС и пр.
Прогноз синдрома Элерса-Данлоса
На качество и продолжительность жизни больных синдромом Элерса-Данлоса влияет тип заболевания. Наиболее серьезный прогноз имеет IV тип синдрома Элерса-Данлоса – летальный исход может наступить вследствие разрывов сосудов, внутренних органов и кровотечений. Наличие синдрома I типа существенно ограничивает качество жизни. Относительно благоприятно протекание II—III типов болезни.
В целом, наличие синдрома Элерса-Данлоса сопряжено со множеством социальных трудностей, ограничивает полноценную физическую активность и выбор профессии.
Синдром Элерса-Данло
Синдром Элерса-Данло (Ehlers, Danlos) (СЭД) – это полиморфная группа заболеваний, обусловленных наследственной патологией соединительной ткани (мезенхимальной дисплазией). Синонимы: гиперэластическая кожа, “каучуковый человек”.
Заболевание встречается в практике врачей разных специальностей. В настоящее время выделены 11 типов проявлений СЭД, которые различаются по типу наследования, по распространенности дефектов, клиническим и морфологическим проявлениям, по биохимическим характеристикам. У каждого из типов СЭД имеются свои особенности распределения коллагена III типа, а также актина в коже и в стенках сосудов. Аномальный коллаген находят в соединительной ткани внутренних органов, в связках. У больных СЭД I-III типов обнаруживают истончение рогового слоя и разрыхление базальной мембраны эпидермиса, аномальное строение сетчатого слоя дермы, беспорядочное расположение коллагеновых и эластических волокон. Стенки сосудов истончены. Эти особенности объясняют клинические проявления болезни, ее чрезвычайный полиморфизм. Лучше других описаны и охарактеризованы 8 типов синдрома.
I тип характеризуется генерализованным и тяжелым переразгибанием суставов и гиперэластозом кожи. Тип наследования аутосомно-доминантный. Болезнь чаще встречается у мужчин. Начинается обычно с детских лет. В первую очередь обращает внимание гиперэластичная кожа и ее легкая ранимость, когда даже незначительные травмы приводят к возникновению зияющих ран, заживающих с образованием атрофических рубцов, похожих на папиросную бумагу, на лбу, локтях, щеках, на коленях.
Ушибы и другие травмы кожи могут приводить к быстрому образованию подкожных гематом. У больных очень плохо заживают послеоперационные раны (часты расхождения и прорезывание швов, послеоперационные грыжи и т.п.); у женщин происходит преждевременный разрыв околоплодных оболочек, что служит причиной недонашивания беременности.
Больным свойственна чрезмерная подвижность и разболтанность суставов. Это приводит к частым подвывихам и вывихам суставов пальцев кистей и стоп. Но парадоксально то, что именно эти особенности позволяют некоторым индивидам с СЭД добиваться значительных успехов при работе в цирке, балете.
Могут обнаруживаться и другие пороки развития: кифосколиоз, вдавление грудины, широкая переносица, выступающие лобные бугры, хондродистрофия, синдактилия, плоскостопие, дефекты глаз, эпикантус. Встречаются бронхоэктазы, висцероптоз, диафрагмальная грыжа, варикозное расширение подкожных вен, гиперпигментации. Нередки нарушения интеллекта. Описаны нарушения формы зубов и мест их промывания. У некоторых больных обнаруживают моллюскоподобные псевдоопухоли и подкожные шарики.
При І, II и V типах СЭД часто обнаруживают признаки поражения ССС: пролапс митрального клапана, врожденные пороки сердца, аневризму аорты и других сосудов. Наблюдаются кардиалгии, различные нарушения проводимости и ритма.
У больных СЭД нередко находят на передней поверхности голеней единичные плотные подкожные узелки размерами от 2 до 5 мм, состоящие из скопления жировых клеток с плотной фиброзной капсулой; в них могут образоваться кальцификаты. Реже узелки и моллюскоподобные псевдоопухоли находят на локтях и в области коленных суставов. Периодически может возникать невысокая лихорадка.
2 тип СЭД характеризуется менее выраженными изменениями. Повышенная подвижность суставов может обнаружиться, например, только в пальцах кистей и стоп. Кожные изменения минимальны, но бывают кровоподтеки, а рубцы после травм образуются плохо.
3 тип СЭД, доброкачественный. Выраженная гипермобильность и разболтанность всех суставов, обычно без мышечно-скелетных деформаций. Кожные изменения минимальны.
4 тип СЭД, так называемый артериальный или экхимозный, наиболее злокачественный. Выраженная склонность к спонтанному разрыву крупных и средних артерий и к перфорации кишечника, преимущественно сигмовидной кишки. Кожа у этих больных очень тонкая, но слаборастяжимая, с подчеркнутым рисунком поверхностных вен. В генезе этого типа СЭД имеет значение сниженный синтез коллагена III типа. Аутосомно-рецессивный тип наследования.
5 тип СЭД проявляется только повышенной растяжимостью кожи. Подвижность суставов минимальная. Умеренная склонность к возникновению кровоподтеков. Невыраженная хрупкость кожи. Первичным дефектом при этом типе СЭД является дефицит лизилоксидазы. Встречается только у мальчиков, так как наследование идет по рецессивному, сцепленному с Х-хромосомой, варианту.
6 тип СЭД – глазной, характеризуется “глазной хрупкостью” и тяжелым сколиозом в сочетании с небольшими изменениями кожи и суставов. При этом типе СЭД разрывы склеры и роговицы, отслойка сетчатки глаза возможны даже вследствие легчайшей травмы. Первичным дефектом здесь выступает недостаточность лизилгидроксилазы, при аутосомно-рецессивном типе наследования.
7 тип СЭД представлен генерализованной подвижностью суставов при низком росте. У больных часты подвывихи бедра, коленных суставов, стоп. Умеренные растяжимость кожи и склонность к кровоизлияниям. На лице выражены гипертелоризм и эпикант. Первичный дефект при этом кроется в дефиците проколлагеновой пептидазы. Аутосомно-рецессивный тип наследования.
8 тип СЭД заключается в жировом некробиозе кожи диабетического происхождения. Выражен периодонтоз с преждевременным выпадением зубов вследствие рассасывания альвеолярного ложа. На коленных суставах много мелких рубцов, подобных папиросной бумаге. Подвижность суставов вариабельна, Аутосомно-доминантный тип наследования.
Другие типы СЭД менее обозначены с клинической точки зрения. Дифференциальная диагностика СЭД в первую очередь необходима с другими вариантами мезенхиальной дисплазии: с синдрома Марфана, с так называемой вялой кожей, когда она, свисая складками, не обладает растяжимостью. Гипермобильность суставов известна при синдроме Ашара (см. ниже).
В дополнение к типичной клинической картине, соответствующей конкретному типу СЭД, проводится тест па подвижность пальцев. Обычно положительны тесты разгибания мизинца на 90° и более и активного приведения 1-го пальца кисти к предплечью.
Диагностика.
Морфологическая картина СЭД при биопсии кожи выявляет обычно картину увеличения эластических волокон и уменьшение коллагеновой структуры, но электронно-микроскопические изменения коллагеновых фибрилл неспецифичны. Типы СЭД могут быть уточнены при углубленном обследовании клиническим генетиком. Однако легкие варианты СЭД часто остаются незамеченными врачом.



