Что такое системные липоидозы
Липидозы — заболевания, характеризующиеся аномалийным накоплением сфинголипидов в макрофагах костного мозга и других тканей. Эти болезни известны в патологии под различными названиями: тезаурисмозы, гистиомоноцитарные липидозы, метаболические или аккумулятивные ретикулезы.
Патогенез липидозов. Главное расстройство, стоящее в основе всех липидозов, состоит в неспособности деградации сфинголипидов и их накоплении в макрофагах различных тканей.
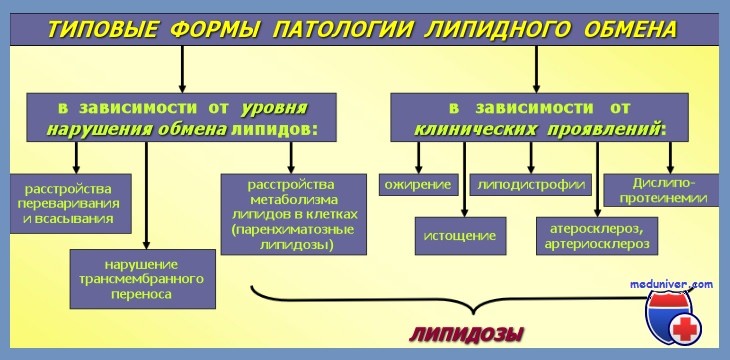
Главный компонент различных сфинголипидов — церамид, который является ацилированным сфингозином. Она выполняет важнейшие структурные функции на уровне многих клеток в организме. Жирнокислотная часть сфинголипидов на уровне мозга представлена в особенности стеариновой кислотой (С18), а на уровне остальных тканей жирными кислотами с высоким числом углеродов (С20—С21). Отличительная черта и функция каждого сфинголипида определяются радикалом, с которым связан церамид.
Большинство этих болезней возникают из-за врожденных энзиматических дефицитов: наследственные липидозы. При этом типе заболеваний накопление сфинголипидов происходит не из-за сверхпроизводства липидов, а из-за отсутствия или сокращения некоторых специфических гидролитических энзим (Brady, Turpin).
В других случаях не существует энзиматического дефицита, а накопление липидов происходит благодаря чрезмерному разрушению клеток (гранулоцитов, эритроцитов, тромбоцитов), которые высвобождают большие количества липидного материала — приобретенные или вторичные липидозы (Albrecht, Dosik, Kattlowe, Silverstein, Ursea, Zaino).
Независимо от механизма тезауризации, наиболее затронутыми тканями оказываются те, которые представляют повышенный турновер липидов: развивающаяся нервная система и моноцитомакрофаговая система, где, в результате фагоцитоза, катаболизм липидов составляет главную функцию этих клеток.

Изучение липидозов имеет значение для гематологов из следующих соображений:
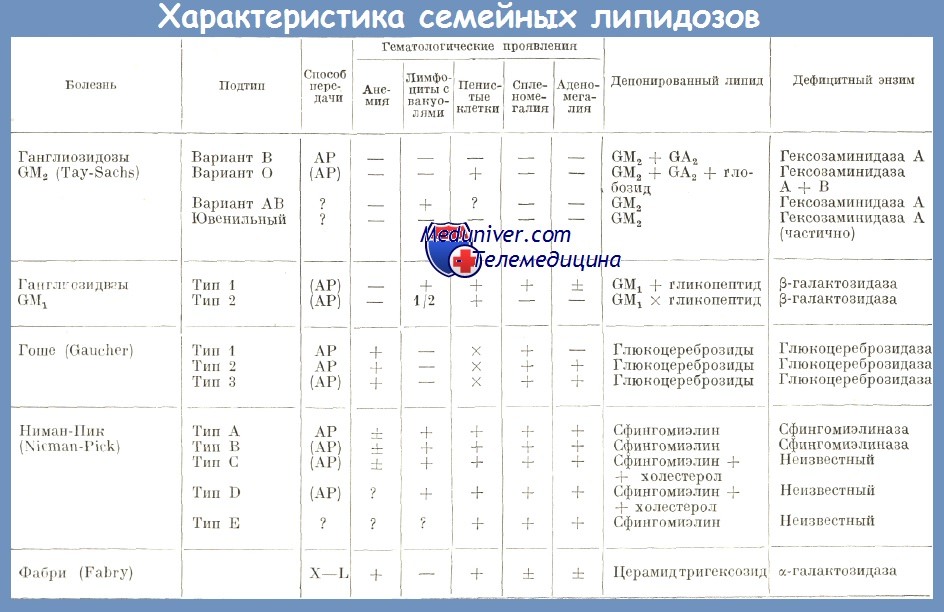
а) некоторые болезни представляют гематологические явления (болезнь Гоше, Нимана-Пика); другие представляют кожные поражения петехиального типа (болезнь Фабри);
б) липиды, накопляющиеся в макрофагах, происходят от эритроцитов, гранулоцитов и тромбоцитов;
в) в течение гематологических заболеваний могут появляться вторичные липидозы.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Липоидозы симптомы и лечение
Липоидозы – составляют большую группу аномалий липидного обмена, носящих наследственный характер, объединенных высоким уровнем содержания липидов в плазме крови или накоплением метаболитов внутриклеточного липидного обмена. Среди внутриклеточных липоидозов, или так называемых болезней накопления, следует назвать в первую очередь болезнь Гоше, иначе глюкоцереброзидоз, болезнь Ниммана-Пика, иначе сфингомиелиноз, амвротическую идиотию, иначе цереброретинальная дегенерация, и др. При плазматической природе липоидозов присутствует гиперлипидемия, представляющая фактор риска развития атеросклероза, панкреатита и т.д.
Симптомы липоидозов
Лечение липоидозов
Лечение липоидозов нашими специалистами проводится после определения точного диагноза путем тщательного обследования пациента с применением самых современных методов. Стратегия лечения включает разработку медицинских мероприятий, направленных на достижение сбалансированности липидного обмена и предупреждение негативных ситуаций сопутствующих развитию болезни. В зависимости от вида заболевания могут быть подключены физиотерапевтические процедуры. Опытные врачи дадут необходимые рекомендации по рациональному питанию.
Консультация и прием врача
Более подробную информацию Вы можете узнать по телефонам указанным на сайте или обратиться в наш Медицинский Центр. Мы работаем Без Выходных с 8.00 до 22.00 по адресу: г.Москва ВАО (Восточный Административный Округ) Сиреневый Бульвар 32А
Липоидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов и их метаболитов в различных органах и тканях. Общие клинические проявления представлены прогрессирующим расстройством интеллектуальных и двигательных функций, поражением костей, кожи, центральной нервной системы, глаз и внутренних органов (печени, почек, селезенки). Диагностика основана на лабораторных исследованиях ферментативной активности, количества токсичного субстрата, наличия мутации в генах. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
МКБ-10
Общие сведения
Синонимичные названия липоидозов – липидозы, ретикулоэндотелиозы, лизосомные болезни накопления липидов. К данной группе относится сфингомиелиноз, болезнь Тея-Сакса, болезнь Гоше, семейные гиперлипидемии и некоторые другие заболевания. Общей характеристикой является патологическое накопление липидов внутри клеток организма в результате дефекта ферментных систем. Липоидозы относятся к группе редких (орфанных) заболеваний. Их распространенность очень низка, для отдельных типов патологий составляет от 1:40 тыс. до 1:1 млн. и реже. Суммарная частота – 1:7 тыс. Большинство липидозов имеют прогредиентное течение, приводят к инвалидизации и ранней смерти.
Причины липоидозов
Определяющим фактором развития липидозов является генетический дефект, который обуславливает полную или частичную недостаточность лизосомальных ферментов, расщепляющих сложные липиды. Наследование болезней происходит по аутосомно-рецессивному механизму. Это означает, что новорожденный оказывается больным, если получает мутационный ген от каждого родителя (имеет пару дефектных генов в аллели). Когда мутация передается только от матери или отца, то ребенок является ее носителем и остается здоровым.
Исключительный механизм передачи дефекта у болезни Фабри. В отличие от других липидозов, она наследуется по X-сцепленному рецессивному типу. Гемизиготные пациенты мужского пола больны, передают мутацию только дочерям. У девочек заболевание всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза проявляются у пациенток с одним дефектным геном, когда доминантный ген оказывается инактивированным (причины этого не выяснены).
Патогенез
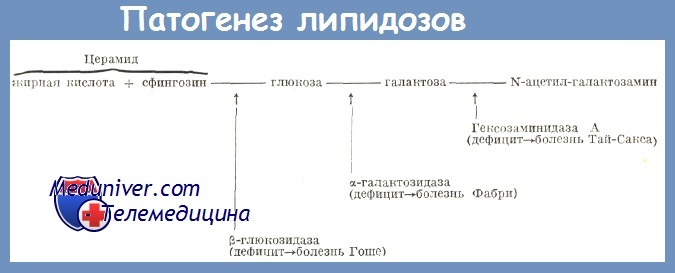
Патогенетической основой большинства липоидозов является генетически детерминированный ферментативный дефект – энзимопатия. Вследствие этого лизосомы клеток накапливают липиды и промежуточные продукты их метаболизма, что ведет к прогрессивному нарастанию нарушения функций органов. При синдроме Вольмана определяется недостаточность кислой этеразы, становится невозможным полный цикл обмена холестерола, повышается содержание его эфиров в лизосомах селезенки, печени, надпочечников, кишечника и костного мозга. У пациентов с болезнью Гоше снижено или полностью отсутствует производство бета-глюкозидазы; в печени, селезенке, костном мозге скапливаются продукты расщепления сфинголипидов. Болезнь Нимана-Пика развивается на базе дефицита сфингомиелазы, характеризуется повышением количества сфингомиелина во многих клетках, особенно в гепатоцитах, нейронах. При идиотии Тэя-Сакса существует дефект N-ацетилгексозаминидазы, происходит накапливание ганглиозидов в головном мозге.
Классификация
Наследственные нарушения метаболизма сложных липидов представлены группой различных по происхождению заболеваний, которые на патогенетическом уровне связаны с патологией жирового обмена. Для липоидозов характерно скопление сложных липидных соединений внутри лизосом клеток. В зависимости от того, какой липид не расщепляется до конца и откладывается в тканях, выделяют несколько типов болезней:
Симптомы липоидозов
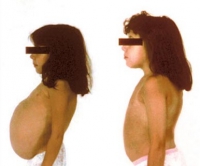
Клиническая картина липидозов определяется особенностями вовлечения в патологический процесс органов и систем. При болезни Гоше I типа поражается печень, селезенка, кости и костный мозг. Симптомы включают увеличение размеров печени, хрупкость костей, анемию, лейкопению, снижение свертываемости крови. II тип заболевания разворачивается с преимущественным повреждением ЦНС и печени. Наблюдаются судорожные приступы, мышечный гипертонус, спастичность, интеллектуальная недостаточность, расстройства акта глотания. У людей с галактозилцерамидным липидозом (болезнью Краббе) снижается функциональность миелиновой оболочки. Развивается гипервозбудимость, рвота, судороги, задержка психомоторного развития с прогрессирующим снижением интеллекта и зрения.
При метахромотической лейкодистрофии патологически изменяется миелиновая оболочка. Основные симптомы – гипотония мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, глухота, интеллектуальное и моторное недоразвитие. Клинические признаки болезни Андерсона-Фабри разнообразны, одними из наиболее распространенных являются нейропатическая боль (умеренной и легкой выраженности, в ступнях и ладонях) и кризы Фабри – сильные жгучие боли в конечностях приступообразного характера. Дополнительно возникает гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератомы, расстройства слуха, сердечно-сосудистой и почечной функций.
При развитии синдрома Сандхоффа определяется общая вялость, гипотонус мышц конечностей, трудности сосания и глотания, прогрессирующая задержка моторного и психического развития, двигательная слабость, шумы в сердце, судороги, слепота, увеличение селезенки. При ранней детской амавротической идиотии больше всего поражается центральная нервная система. К концу первого полугодия ухудшается реакция детей на внешние сигналы (лица близких, игрушки), утрачиваются двигательные навыки, снижается познавательный и игровой интерес. Нарушается зрение вплоть до слепоты. Формируются судорожные припадки.
Первые симптомы сфингомиелиноза – вялость, малоподвижность, апатичность, отказ от еды, рвота. Позже увеличивается живот (гепатомегалия), конечности становятся худыми, кожа приобретает коричневатый оттенок, периоды заторможенности сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Диагностируется умеренная гидроцефалия, гипертермия, спастический парез ног и рук, приступообразная асфиксия.
Проявления гиперлипопротеинемий обнаруживаются в возрасте до 10 лет. Для всех форм болезней свойственно отложение подкожного жира в виде ксантом, а также абдоминальные боли, панкреатит, гепатоспленомегалия (скопление липидов в селезенке, печени). При комбинированной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз сосудов. При гиперлипидемиях IV и V типа – снижение чувствительности к глюкозе, ИБС.
Осложнения
Липоидозы протекают с развитием недостаточности жизненно важных органов. Наиболее частыми осложнениями становятся заболевания сердца и сосудов, ЦНС, почек, легких, печени. Пациенты страдают от атеросклероза сосудов, сердечной и дыхательной недостаточности, ишемической болезни сердца, хронической почечной и печеночной недостаточности, аденом и цирроза печени, инсультов, транзиторных ишемических атак. При типах заболеваний, совместимых с жизнью, зачастую нарушено двигательное и психическое развитие. Многие больные оказываются неспособны к самообслуживанию, обучению и овладению профессией, нуждаются в пожизненном уходе со стороны окружающих.
Диагностика
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением занимаются педиатры, гематологи, гастроэнтерологи, ревматологи, неврологи, психиатры и врачи-генетики. На первичном этапе диагностики проводится сбор семейного анамнеза: при наследственном характере ферментопатии у больного могут быть родственники с подтвержденным диагнозом липидоза. При сборе клинических данных специалисты обращают внимание на время начала симптомов: чаще всего болезнь проявляется в период новорожденности или первого года жизни, редко – у детей постарше или взрослых. К специфическим методам обследования пациентов относят:
Лечение липоидозов
Терапия данной группы заболеваний – сложная задача для врачей разных специальностей. Методы лечения несовершенны и продолжают разрабатываться, при некоторых видах болезней возможно добиться лишь временного улучшения самочувствия больного, при других достижима стойкая ремиссия. Общая схема медицинской помощи больным состоит из трех компонентов:
Прогноз и профилактика
Липоидозы характеризуются гетерогенностью генетического дефекта и выраженной клинической полиморфностью. Большинство из них имеет непрерывное прогрессирующее течение. Правильная диагностика и своевременное лечение позволяют увеличить продолжительность жизни больных, а при легких формах способствуют улучшению адаптации. Профилактика возможна на этапе планирования и в первые месяцы беременности. Супружеским парам с высоким риском передачи болезни ребенку рекомендуется медико-генетическое консультирование, а в первом триместре – исследование амниотической жидкости и материала биопсии хориона на наличие мутаций генов.
Липидозы
Полезное
Смотреть что такое «Липидозы» в других словарях:
Дистрофия клеток и тканей — нарушение тканевого или клеточного обмена, сопровождающееся определенными структурными изменениями клеток и межклеточного вещества. В основе развития дистрофии лежат расстройства регуляторных механизмов трофики врожденного или приобретенного… … Медицинская энциклопедия
Кожа — I Кожа (cutis) сложный орган, являющийся наружным покровом тела животных и человека, выполняющий разнообразные физиологические функции. АНАТОМИЯ И ГИСТОЛОГИЯ У человека площадь поверхности К. равна 1,5 2 м2 (в зависимости от роста, пола,… … Медицинская энциклопедия
Дистрофия — (от Дис. и греч. trophe питание) дегенерация, перерождение, патологический процесс, возникающий в связи с нарушением обмена веществ и характеризующийся появлением в тканях продуктов обмена веществ, изменённых количественно и качественно … Большая советская энциклопедия
Молекулярные болезни — («Молекулярные болезни», ) врождённые ошибки метаболизма, заболевания, обусловленные наследственными нарушениями обмена веществ. Термин «М. б.» предложен американским химиком Л. Полингом. В начале 20 в. английский врач Л. Э. Гаррод,… … Большая советская энциклопедия
Гепатолиена́льный синдро́м — (греч. hēpar, hēpat[os] печень + лат. lien селезенка; синоним печеночно селезеночный синдром) сочетанное увеличение печени (гепатомегалия) и селезенки (спленомегалия), обусловленное вовлечением в патологический процесс обоих органов. Встречается… … Медицинская энциклопедия
Гликогено́зы — (glycogenosis, единственное число; гликоген + sis; синоним: болезнь накопления гликогена, гликогеновая болезнь) группа наследственных болезней, которые обусловлены недостаточностью ферментов, участвующих в обмене гликогена; характеризуются… … Медицинская энциклопедия
Гоше болезнь — I Гоше болезнь (Ph.Ch.Е. Gaucher, франц. дерматолог, 1854 1918) наследственная болезнь, характеризующаяся накоплением глюкоцереброзидов в макрофагах главным образом селезенки, костей и печени; наследуется по аутосомно доминантному типу см.… … Медицинская энциклопедия
Жирово́й обме́н — совокупность процессов переваривания и всасывания нейтральных жиров (триглицеридов) и продуктов их распада в желудочно кишечном тракте, промежуточного обмена жиров и жирных кислот и выведение жиров, а также продуктов их обмена из организма.… … Медицинская энциклопедия
Липиды — I Липиды (греч. lipos жир + eidos вид) класс жиров и жироподобных веществ (липоидов), различающихся по химическому составу, структуре и выполняемым в организме функциям, но сходных по физико химическим свойствам. Все Л. нерастворимы в воде, но… … Медицинская энциклопедия
Ниманна — Пика болезнь — I Ниманна Пика болезнь (A. Niemann, нем. педиатр, 1880 1921; L. Pick, нем. патолог, 1868 1944) наследственный липидоз, в основе развития которого лежит избыточное накопление сфингомиелина в клетках системы мононуклеарных фагоцитов и ц.н.с., см.… … Медицинская энциклопедия
Липидозы
Группа заболеваний, характеризующихся нарушением липидного обмена и имеющих преимущественно наследственный характер.
Болезни накопления, которые обусловлены отложением аномально больших количеств нерасщепленных продуктов жирового обмена в различных органах и тканях, что приводит к значительному нарушению их функции.
Что провоцирует
Патогенез липидозов
Диагностика
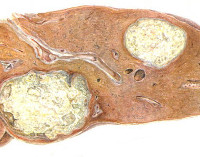
Морфологические исследования направлены на выявление специфических признаков патологического накопления макромолекул в клетках костного мозга, селезенки, нервной ткани, фибробластов кожи. В ряде случаев эти изменения очень типичны (клетки Гоше, Нимана-Пика).
Болезни накопления
Болезнь Тея-Сакса
Патогенез
В патогенезе ведущая роль отводится дефициту фермента бета-гексозаминидазы А в плазме крови, мозге и внутренних органах, что приводит к накоплению в их клетках ганглиозида Gm2.
Проявление болезни
Клинический диагноз амавротической идиотии Тея-Сакса
Педположение о болезни Тея-Сакса возникает, когда, наряду с терапевтическим осмотром, окулист (врач, специализирующийся на заболеваниях органов зрения) обнаруживает на глазном дне вишнево-красное пятно.
Определение количества фермента в жидкостях и тканях больного необходимо для подтверждения диагноза или для выявления носительства.
Необходимы анализ крови и биопсия кожи (анализ крошечных срезов кожи).
Пренатальная диагностика возможна при амниоцентезе (аспирация и анализ амниотической жидкости, полученной проколом плодного пузыря).
Клинический диагноз амавротической идиотии Тея-Сакса основывается на:
Лечение
Болезнь Тея-Сакса не поддается лечению. Нарастающая клиническая картина заболевания, угасающий больной ребенок мучительны для родных и близких. Здесь может помочь только участие и эмоциональная поддержка.
Болезнь Ниманна — Пика
Болезнь Нимана — Пика (ретикулогистиоцитарный сфингомиелиноз, фосфатидный липидоз)
Патогенез
Формы
Ранняя тяжелая
Поздняя хроническая
Типы
Заболевание встречается во всех этнических группах, однако частота типа А выше среди евреев-ашкенази и составляет 1: 100.
Тип А (классическая инфантильная форма, острая нейропатическая форма) составляет более половины всех случаев болезни Нимана-Пика.
Заболевание проявляется после рождения и характеризуется поражением внутренних органов и ц.н.с.
Клиническая картина (тип А):
Уже в 3 месяца отмечаются трудности вскармливания, гипотрофия, а в 6 месяцев выявляется гепатоспленомегалия.
Как правило, печень увеличивается раньше, чем селезенка.
Дети истощены, характерны большой выступающий живот и тонкие конечности.
Из неврологических нарушений отмечаются мышечная гипотония, угнетение сухожильных рефлексов, отсутствие реакции на окружающее, остановка моторного развития, затем утрата уже приобретенных навыков.
Рано снижается слух. Кожа приобретает коричневато-желтую окраску из-за нарушения обмена сфингомиелина.
Примерно в 50% случаев выявляется вишнево-красное пятно в области желтого пятна сетчатки.
Также описаны помутнение роговицы, коричневое прокрашивание передней капсулы хрусталика.
Больные дети умирают обычно на третьем году жизни.
При типе В (висцеральная форма, хроническая форма без вовлечения нервной системы) основные клинические проявления развиваются позже, чем при типе А.
Спленомегалия появляется в возрасте 2-6 лет, позднее поражаются печень и легкие (больные подвержены частым инфекциям дыхательных путей).
Симптоматика поражения ц.н.с. отсутствует, напротив, в ряде случаев отмечены высокие интеллектуальные способности.
Продолжительность жизни не снижена.
Тип С (подострая, юношеская форма, хроническая нейропатическая форма) проявляется в 1-2 года и характеризуется нейровисцеральными нарушениями.
Сначала появляется гепатоспленомегалия (менее выражена по сравнению с типами А и В), может наблюдаться холестаз.
Неврологические симптомы развиваются на фоне поражения внутренних органов, отмечаются мышечная гипотония, повышение глубоких сухожильных рефлексов, которые сменяются спастическим параличом, а также интенционный тремор, умеренная атаксия, судороги.
Большинство больных погибает в возрасте 5-15 лет.
Диагноз болезни Ниманна-Пика основывается на данных:
Лабораторная диагностика
Лечение
Болезнь Гоше (глюкозилцерамидный липидоз)
Этиология, патогенез
Классификация
Тип 1 (доброкачественный) в 30 раз чаще встречается у евреев (западноевропейской группы Ашкенази); неврологические нарушения при этом отсутствуют, висцеральные изменения связаны преимущественно с кроветворными органами, увеличением селезенки, явлениями гиперспленизма, деструкцией костной ткани. При двух других типах какого-либо этнического преобладания не отмечено.
Тип 2 представляет собой злокачественную форму процесса с грубыми неврологическими нарушениями, которые проявляются уже у новорожденных и ведут к смерти в первые 2 года жизни.
Тип 3 отличается вариабельностью висцеральных и неврологических нарушений; по течению он менее злокачествен, чем тип 2
Хроническая форма встречается у детей любого возраста, но чаще после 5-8 лет.
Рано увеличивается живот (спленомегалия). Возникают спонтанные боли в ногах (рентгенологически: остеопороз либо остеосклероз). Возможна колбообразная деформация бедра по типу «бутылки». Кожа лица, шеи, ладоней и стоп коричневая, с охряно-желтым или бронзовым оттенком, пигментация может перейти в диффузную и захватить слизистые оболочки; кроме того, бывают различных размеров и очертаний кровоизлияния. Возможны носовые и кишечные кровотечения. В крови: анемия, лейкопения, гранулоцитопения, тромбоцитопения; уровень холестерина и липидов в пределах нормы, повышены активность кислой фосфатазы и содержание р-глобулинов. Общее состояние ребенка долго остается удовлетворительным, но постепенно развивается отсталость физического развития, все проявления болезни прогрессируют, нарастает анемизация, снижается иммунитет.
Прогноз определяется возрастом: он тем тяжелее, чем младше ребенок.
Диагноз
Лечение
С выделением и очисткой глюкоцереброзидазы стало возможным замещение мутантного фермента у больных. Десятилетний опыт применения ферментозаместительной терапии во всем мире свидетельствует о том, что этот метод лечения останавливает прогрессирование заболевания, способствует обратному развитию симптомов болезни Гоше и значительно улучшает качество жизни больных. Сегодня во всем мире тысячи больных с болезнью Гоше получают внутривенные инъекции модифицированной человеческой глюкоцереброзидазы.
В Украине двум детям проводится специфическое лечение в качестве гуманитарной помощи фирмы Джензайм.
Благодаря успехам генетики и современной медицинской науки эффективное лечение наследственных болезней становится реальностью, уменьшает боль и страдания многих людей, дарит ранее безнадежным пациентам надежду на полноценную жизнь.
Марцинковская И.Р.
5 курс 1 мед 2 группа



