Что такое время полувыведения препарата
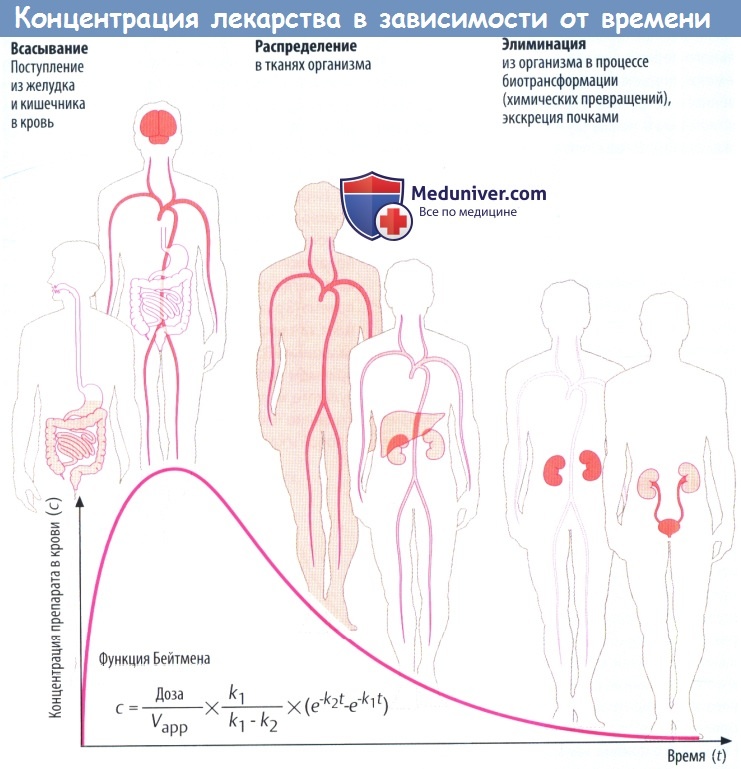
Такие процессы, как всасывание и выведение, обладают экспоненциальными характеристиками. В отношении всасывания это следует из простого факта: количество препарата, перемещающегося за единицу времени, зависит от разности концентраций (градиента) на границе двух тканей (закон Фика).
В процессе всасывания из пищеварительного тракта содержимое кишечника и кровь представляют собой ткани с изначально высокой и низкой концентрациями соответственно. При выведении лекарственного вещества через почки экскреция часто зависит от скорости клубочковой фильтрации, т. е. от количества препарата, попавшего в первичную мочу.
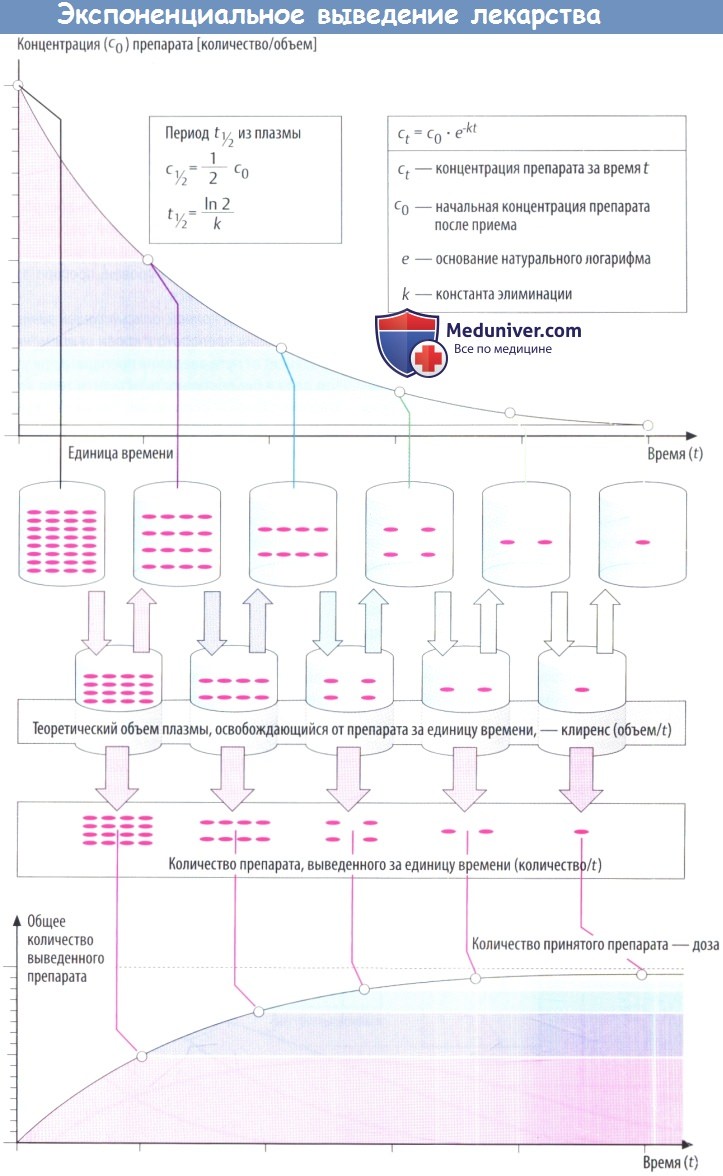
По мере снижения концентрации в крови количество лекарственного вещества, экскретируемого через почки за единицу времени, уменьшается. В результате происходит экспоненциальное снижение, показанное на рисунке ниже. Время экспоненциальногоснижения — постоянный интервал, в течение которого концентрация снижается в 2 раза.
Этот интервал представляет собой период полувыведения (t1/2) и связан с константой скорости элиминации (k) уравнением: t1/2 = (ln2)/k. Эти два параметра вместе с исходной концентрацией (с0) описывают скорость реакции первого порядка (экспоненциальную).
Поскольку эта скорость постоянная, она дает возможность вычислить объем плазмы, освобожденной от лекарственного вещества, учитывая, что оставшееся количество не распределено равномерно в общем объеме плазмы (условие, невозможное в реальности). Теоретический объем плазмы, освобождающейся от лекарственного вещества за единицу времени, называется клиренсом.
В зависимости от того, снижается концентрация в плазме в результате экскреции с мочой либо в результате разрушения в процессе метаболизма, клиренс называют почечным или печеночным. Почечный и печеночный клиренсы суммируются, образуя общий клиренс (Cltot) в случае, если препараты выводятся в неизмененном виде через почки и подвергаются биотрансформации в печени.
Cltot представляет собой сумму всех процессов, участвующих в выведении; он связан с периодом полувыведения (t1/2) и объемом распределения препарата (Vapp) формулой:
Чем меньше объем распределения и чем больше общий клиренс, тем короче период полувыведения.
Для препаратов, выводимых почками в неизмененном виде, t1/2 можно вычислить на основании кумулятивной экскреции с мочой; итоговое общее количество выведенного препарата соответствует количеству всосавшегося препарата.
Печеночная элиминация происходит по экспоненте, т. к. ферменты, катализирующие реакции метаболизма, действуют в квазилинейной области своей кривой активности концентрации; следовательно, количество вещества, подвергшегося метаболизму за единицу времени, уменьшается параллельно снижению концентрации в крови.
Самое известное исключение из экспоненциального закона — выведение алкоголя (этанола), которое происходит по линейному закону (кинетика нулевого порядка), во всяком случае, при концентрации в крови менее 0,02%. Это происходит потому, что лимитирующий скорость фермент алкогольдегидрогеназа достигает полунасыщения при очень низких концентрациях вещества — примерно 80 мг/л (0,008%).
Таким образом, при концентрации этанола в крови на уровне примерно 0,02% скорость реакции выходит на плато, при концентрациях выше этого уровня количество лекарственного вещества, выведенного за единицу времени, остается постоянным.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Что такое время полувыведения препарата
В распределении анестетика важную роль играет также связывание с белками плазмы, наибольшее значение из которых имеют альбумины. Связанная с белками часть препарата образует депо и находится в равновесии с растворенной в плазме частью, но лишь растворенная часть препарата (не связанная с белками плазмы) распределяется в тканях и оказывает фармакологическое действие. Различные прераты при одновременном введении вместе конкурируют за места связывания с белками плазмы.
В результате концентрация свободной фракции отдельных препаратов в плазме может возрасти, что проявляется признаками передозировки. Аналогичный эффект может вызвать уменьшение белково-связывающей емкости плазмы при заболеваниях печени и почек, а также алиментарной белковой недостаточности. В этих случаях дозу анестетика можно уменьшить. Еше большее практическое значение имеет зависимость связывания препарата с белками плазмы от скорости его введения.
При быстром введение препарата свободная его фракция (т.е. экологически активная часть) увеличиваются. Во избежание острой передозировки внутривенные анестетики вводят можно и с учетом достигаемого эффекта.|
Элиминация. Внутривенные анестетики подвергаются биотрансформации. Отчасти метаболизируются или инактивируются в печени и выводятся с желчью, почками (гепаторенальный клиренс). Лишь небольшая часть препаратов выдится из организма в неизмененном виде. Метаболизация с участием фермнтов более длительный процесс, чем вывод через легкие. Поэтому время элиминации даже современных внутривенных аанестетиков короткого действия больше ингаляционных анестетиков.
Период полувыведення. При системном применении фармакологических препаратов различают три объема (пространства, камеры) распределения: 1. Плазма крови, составляющая 4 объема тела (центральный объем).
2. Интерстициальное пространство (15%).
3. Внутриклеточный объем (40%). Поскольку эндотелий большинства органов содержит межклеточные поры или фенестрирован, то проникновение веществ через него происходит относительно беспрепятственно и зависит только от размера молекул. В связи с этим представляется привлекательной концепция, согласной которой интерстициальное пространство и плазма с точки зрения фармакокинетики рассматриваются как единое (внеклеточное) пространство.
Для активного проникновения внутривенного анестетика решающее значение имеет скорость, с которой анестетик диффундирует из центрального объема (плазма крови) в более глубокие пространства головного мозга. Математически этот процесс можно описать уравнением, рассчитав равновесный период полувыведения 11/2 keO. Он позволяет судить о начале действия препарата. Время, в течение которого происходит начальное распределение препарата по всему организму, обозначают как период полураспределения (tl/2a).
После завершения распределения между концентрациями препарата в отдельных пространствах устанавливается устойчивое равновесие (steady state). Дальнейшая динамика концентрации определяется в первую очередь процессом элиминации анестетика (клиренс плазмы). Время, необходимое для уменьшения концентрации вещества в плазме до половины исходного уровня, называют периодом полувыведения (tl/2p). Снижение концентрации, как правило, описывается логарифмической зависимостью.
Период полувыведения вещества не следует отождествлять с длительностью его действия (см. выше)! Чтобы рассчитать элиминацию при непрерывном поступлении препарата (ТВА, ИУЦК), основываются на контекстно-чувствительном периоде полувыведения. Под контекстно-чувствительным периодом полувыведения понимают время, за которое концентрация анестетика в плазме крови после прекращения внутривенной его инфузии уменьшается на 50%. Элиминационный период полувыведения анестетика определяют исходя из его плазменного клиренса и объема распределения. Этот период тем короче, чем больше клиренс и чем меньше объем распределения.
Период выведения и время полураспада лекарств

После всасывания в кровь лекарственные средства (ЛС) неравномерно распределяются в органах и тканях организма. Существенно влияют на распространение веществ биобарьеры. К ним относятся стенка капилляров, цитоплазматический, гематоэнцефалический (ГЭБ) и плацентарный барьеры.
Биологические барьеры организма
Большинство препаратов легко преодолевает стенку капилляров. Одни средства проникают через поры путем фильтрации, другие проникают через капиллярную стенку путем диффузии. Некоторые гидрофильные соединения преодолевают капиллярную стенку с помощью транспортных систем.
Выведение лекарств из организма
ЛС и их метаболиты выводятся из организма разнообразными путями: с мочой, калом, желчью, секретом потовых, сальных и бронхиальных желез, молоком матери, воздухом, выдыхаемым воздухом.
Базовую роль в экскреции лекарств играют почки. На выведение лекарств влияют фильтрация, канальцевая реабсорбция и секреция. Фильтрации в клубочках нефрона испытывают вода, глюкоза, аминокислоты, белки с молекулярной массой до 60000 и некоторые другие соединения. Не фильтруются фракции препаратов, связанные с белками плазмы. Скорость фильтрации зависит от интенсивности кровообращения в почках.
В случаях, когда почечный кровоток нарушен (шок, гломерулонефрит и др.), фильтрация существенно уменьшается.
Выделение лекарств с мочой
Активная секреция лекарственных средств происходит в проксимальных отделах нефрона. Секреция из крови через канальцевый эпителий в первичную мочу происходит с затратой энергии с участием специальных транспортных систем. Секретироваться могут как свободные, так и связанные с белками лекарственные средства. Реабсорбция лекарств происходит в дистальных отделах канальцев. Поскольку пассивная реабсорбция происходит через липидные мембраны канальцевого эпителия, то становится очевидным, что лучше реабсорбируются недиссоциированные липофильные молекулы слабых кислот и щелочей, а также нейтральные соединения. Степень реабсорбции зависит от рН мочи. Так, при кислых рН мочи слабые кислоты (барбитураты, бензодиазепины, сульфаниламиды) мало диссоциированные и легко реабсорбируются в кровь.
Выделение лекарств с калом
С калом выводятся из организма препараты, которые плохо всасываются в желудочно-кишечном тракте. Такие препараты используют преимущественно для воздействия на микрофлору кишечника или как слабительные средства.
Некоторые препараты (тетрациклин, пенициллины и др.) выделяются с желчью в тонкий кишечник, откуда они могут выводиться с калом или повторно всасываться, а затем снова выделяться в кишечник (так называемая циркуляция по энтеропеченочную кругу).
Другие способы выведения лекарств из организма
Период полувыведения
Необходимо отметить, что с увеличением дозы препарата выведение его из организма снижается и соответственно возрастает период полувыведения.
Кроме того, для количественной характеристики скорости вывода вещества из организма используют термин «клиренс» (очищение). Он отражает скорость очистки плазмы крови от вещества (например, 10 мл / мин). Различают общий, почечный и печеночный клиренс.
Большинство лекарственных средств несут в организм метаболические изменения. Этот процесс называется биотрансформацией. Суть метаболических превращений заключается в том, чтобы чужеродное, опасное для организма средство превратилось в соединение, которое может быть легко выведено с мочой, желчью или потом. Такие полярные метаболиты плохо растворяются в липидах и имеют низкую способность взаимодействовать с белками плазмы крови и тканей. Метаболиты, как правило, плохо проникают через биологические мембраны и не испытывают реабсорбции в почках и кишечнике.
Метаболизм лекарств в организме
Метаболизм лекарственных средств происходит преимущественно в микросомальном аппарате печени. Некоторые метаболические преобразования определенных лекарств могут происходить в кишечнике, легких, коже и плазме крови. Лишь некоторые препараты выводятся из организма в неизмененном виде.
Известны два базовых вида метаболизма ЛС:
Восстановление является более редким путем метаболизма лекарств. Реакции восстановления катализируют такие ферментные системы, как нитро- и азоредуктазы и др.
Процессы обезвреживания лекарств существенно замедляются у больных с патологией печени (цирроз, острые и хронические гепатиты и др.). Это приводит к росту продолжительности действия препаратов, развития явлений передозировки.
Некоторые препараты могут подавлять микросомальные ферменты печени (левомицетин, бутадион и др.) или немикросомальные ферменты (антихолинэстеразные средства, ингибиторы МАО и др.). В таких случаях действие лекарств, метаболизм которых происходит при участии соответствующих ферментов, увеличивается. В то же время существуют соединения (фенобарбитал и др.), которые повышают (индуцируют) скорость синтеза микросомальных ферментов.
Период полувыведения (полужизни)
Период полувыведения [ править | править код ]
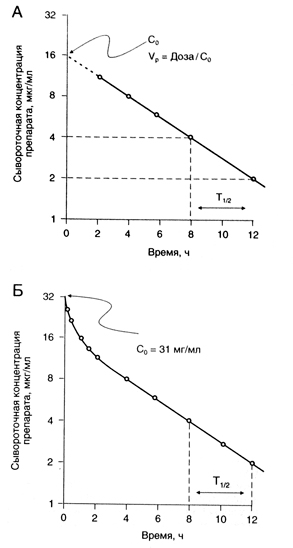
Период полувыведения (Т1/2) — это время, за которое сывороточная концентрация вещества (или его общее содержание в организме) снижается вдвое. В рамках однокамерной модели (рис. 1.4, А) определить Т1/2 очень просто. Полученное значение используют затем для расчета дозы. Однако для многих лекарственных средств приходится использовать многокамерную модель, так как динамика их сывороточной концентрации описывается несколькими экспоненциальными функциями (рис. 1.4, Б). В таких случаях рассчитывают несколько значений Т1/2.
Раньше Т1/2 рассчитывали по участку фармакокинетической кривой, отражающему стадию равновесия (стадию элиминации). С появлением более чувствительных методов измерения концентрации веществ в крови оказалось, что конечный Т1/2 гораздо больше начального. Например, для гентамицина конечный Т1/2 равен 53 ч, тогда как в Приложении II приведен T1/2 2—3 ч. Чрезвычайно длительный конечный Т1/2 индометацина (120 ч) обусловлен, вероятно, активным кишечно-печеночным кругооборотом препарата (в Приложении II приведен Т 1/2 2,4 ч). Клиническая значимость Т1/2 для того или иного периода зависит от того, какая доля вещества выводится из организма и каков объем распределения в этот период, а также от того, какой из показателей — сывороточная концентрация препарата или его общее содержание в организме — лучше коррелирует с фармакологическими эффектами. В Приложении II приведены величины Т1/2, имеющие наибольшее практическое значение.
В прошлом изменение фармакокинетики лекарственных средств при разных патологических состояниях оценивали только на основании Т1/2. В настоящее время общепризнано, что Т1/2 зависит от клиренса и объема рас- пределения вещества. В стационарном состоянии зависимость между Т1/2, клиренсом и объемом распределения приблизительно описывается следующим уравнением:
T1/2 = 0.693 x Vc / Cl(1.12)
Клиренс характеризует способность организма элиминировать вещество, поэтому при снижении этого показателя вследствие какого-либо заболевания Т1/2 должен увеличиваться. Но это справедливо лишь в том случае, если не меняется объем распределения вещества. Например, с возрастом Т1/2 диазепама увеличивается, но не за счет уменьшения клиренса, а вследствие увеличения объема распределения (Klotzetal., 1975). На клиренс и объем распределения влияет степень связывания вещества с белками плазмы и тканей, так что предсказать изменение Т1/2 при том или ином патологическом состоянии не всегда возможно. При остром вирусном гепатите Т1/2 толбутамида уменьшается, а не увеличивается, как это можно было бы ожидать, из-за снижения степени связывания препарата с белками плазмы и тканей. Объем распределения толбутамида не меняется, а клиренс увеличивается вследствие увеличения сывороточной концентрации свободного препарата (Williams et al., 1977).
По Т1/2 не всегда можно судить об изменении элиминации препарата, зато этот показатель позволяет рассчитать время достижения стационарного состояния (в начале лечения, а также при изменении дозы или частоты введения). Сывороточная концентрация, составляющая примерно 94% средней стационарной, достигается за время, равное 4Т1/2. Кроме того, с помощью Т1/2 можно оценить время, необходимое для полного удаления вещества из организма, и рассчитать интервал между введениями (см. ниже).
Стационарное состояние [ править | править код ]
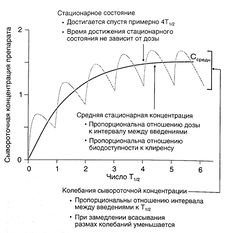
Согласно уравнению 1.1, если вещество вводится путем инфузии с постоянной скоростью, в стационарном состоянии скорость поступления вещества (скорость инфузии) равна скорости его элиминации (произведение клиренса на сывороточную концентрацию препарата — уравнение 1.3). Уравнение 1.1 можно применять и при дробном введении (например, 250 мг каждые 8 ч): в этом случае также устанавливается стационарная сывороточная концентрация препарата, но в промежутках между введениями она колеблется от минимальной до максимальной (рис. 1.5).
Описание к рис. 1.5. Динамика сывороточной концентрации лекарственного средства при дробном введении. Серая кривая описывает накопление препарата при введении с интервалами, равными Т1/2, при условии, что скорость всасывания в 10 раз больше скорости элиминации. При увеличении скорости всасывания максимальная концентрация в стационарном состоянии стремится к 2, а минимальная — к 1. Черная кривая отражает динамику сывороточной концентрации препарата, который вводят в эквивалентной дозе путем инфузии. Обе кривые соответствуют однокамерной фармакокинетической модели. Средняя концентрация в стационарном состоянии вычисляется по уравнению:
Cсредн=F x Доза / (Cl x T)
Это уравнение можно получить путем замены в уравнении 1.1 скорости поступления вещества на выражение F х Доза / Т. Ссредн соответствует концентрации препарата в стационарном состоянии при введении путем инфузии.
Фармакокинетика пероральных антимикробных препаратов
Изучаем особенности фармакокинетики антибактериальных средств в контексте консультирования покупателей аптеки
Абсорбция, распределение и элиминация лекарственного средства тесно связаны с его фармакологическими свойствами и побочными реакциями. Именно поэтому особенности фармакокинетики — предмет пристального интереса как ученых, так и практикующих специалистов. И первостольникам, играющим роль связующего звена между фармацевтической промышленностью, врачом и потребителями, важно помнить, как всасываются и выводятся препараты, включая те, которые отпускаются по рецепту. К тому же разъяснять покупателям особенности приема лекарственных средств, рассказывать о противопоказаниях и побочных эффектах — прямая задача фармспециалиста. И сегодня мы поговорим о фармакокинетике одного из самых обширных и востребованных классов лекарственных препаратов — антибактериальных средств.

Следуя инструкции: общая информация
Прежде всего, рассмотрим самые важные фармакокинетические термины, которые используются в основном в отношении антимикробных средств и часто упоминаются в инструкциях по их применению.
Минимальная ингибирующая концентрация (МИК) представляет собой минимальную концентрацию препарата, которая будет блокировать рост патогенного микроорганизма. Очевидно, что содержание антибиотика в инфицированных тканях должно быть выше, чем МИК. И если препарат А имеет более низкую МИК, чем препарат В, то первый будет убивать возбудителя при более низкой концентрации и, следовательно, проявлять более мощный антибактериальный эффект. Разумеется, при условии, что остальные факторы идентичны [1].
Если уровень МИК препарата для конкретного патогена является низким, последний считается чувствительным к антибиотику. При умеренных значениях МИК чувствительность является промежуточной, а при высоких она отсутствует вовсе, и возбудитель считается устойчивым по отношению к антимикробному средству [1].
Время, в течение которого концентрация препарата в тканях превышает величину МИК (часто обозначается как Т). Антибиотики некоторых групп, в частности, бета-лактамы (ампициллин, амоксициллин и макролиды, за исключением азитромицина) считаются «время-зависимыми» препаратами. Их эффективность определяет концентрация в крови, которая выявляется в течение 40–50 % от длительности интервала дозирования (как правило, около 5–6 часов) [2]. Можно сказать, что эффективность антибиотиков этой категории зависит от продолжительности действия. Чтобы они работали хорошо, их МИК должна быть постоянно превышена.
При уменьшении рекомендуемой кратности приема время-зависимых антибиотиков их эффективность резко снижается [3].
Отношение AUC/МИК — еще один параметр, который часто встречается в инструкциях по применению. Он отражает отношение величины площади под кривой «время-концентрация» (AUC, от англ. аrea under curve) к минимальной ингибирующей концентрации. Считается, что именно AUC/МИК является основным фармакокинетическим параметром эффективности. Препараты, мощность которых определяется как продолжительностью действия, так и концентрацией, — фторхинолоны и тетрациклины [3].
Ну а теперь, вспомнив основные фармакокинетические термины, перейдем к особенностям абсорбции, распределения и выведения современных пероральных антибактериальных ЛС различных групп.
Пенициллины
Основным современным представителем пероральных пенициллинов является амоксициллин. Его наиболее характерные фармакокинетические свойства [1–4]:
Цефалоспорины
Фармакокинетика пероральных и парентеральных цефалоспоринов значительно отличается, что во многом определяет и свойства препаратов, и их показания.
Пероральные цефалоспорины всасываются быстро и хорошо, однако их биодоступность может быть очень разной. Так, биодоступность цефиксима (III поколение) составляет всего 40–50 %. Гораздо более высокие показатели у представителей I и II поколений, таких как цефалексин и цефаклор — до 95 %.
Препаратам этой группы для приема внутрь также свойственны следующие фармакокинетические параметры [1–4]:
Макролиды
Несмотря на то, что все макролиды в основе своей химической структуры имеют макроциклическое лактонное кольцо, их свойства, в том числе и фармакокинетические, значительно разнятся. Особенно выделяется в ряду макролидов азитромицин, содержащий дополнительно молекулу азота в макролидном кольце, что придает последнему повышенную устойчивость [1–4]. Ключевые фармакокинетические свойства марколидов:
Фторхинолоны
Все хинолоны имеют ряд общих фармакокинетических свойств [1–4]:
Тетрациклины
Общие черты фармакокинетики препаратов этой группы выглядит следующим образом [1–4]:
Хлорамфеникол
Также отдельно стоит рассмотреть фармакокинетику хлорамфеникола [1–4]:
Итоги
В заключении хотелось бы подчеркнуть — фармакокинетика часто кажется областью сложной и запутанной. Однако на деле фармакокинетические свойства в пределах одного класса препаратов зачастую схожи, и, выстроив для себя стройный ряд «биодоступность-всасываемость-элиминация» для конкретной группы, можно научиться быстро и легко отвечать на вопросы посетителя о том, когда и как принимать тот или иной антибиотик. Главное — желание и настойчивость, а их провизорам и фармацевтам не занимать.



