«Оптимисты». Как живут люди с орфанными заболеваниями в России?
Ника Воюцкая
Павлику, сыну Снежаны Митиной, было всего три года, когда ему поставили диагноз мукополисахаридоз II типа, или синдром Хантера. «Мы только открыли дверь, а врач уже говорит: „Какой прекрасный Хантер!“ — рассказывает Снежана теперь. — С первой частью я согласилась, а вот кто такой Хантер, мне тогда было неизвестно».
В детском саду мальчик был самым высоким в своей группе, читал стихи про бычка, который идет и качается. Поверить, что совсем скоро он перестанет ходить и говорить, казалось невозможным.
Но в четыре из-за гиперактивности Павлика исключат из детского сада, в шесть — из детского сада для инвалидов: перестанет усваивать программу. У него изменятся внешность и характер.
Терапии, способной корректировать часть симптомов, вместе с другими родителями Снежане придется добиваться самой. Элапразу — единственный препарат, способный помочь ее сыну, зарегистрируют в России не сразу и именно ее трудами.
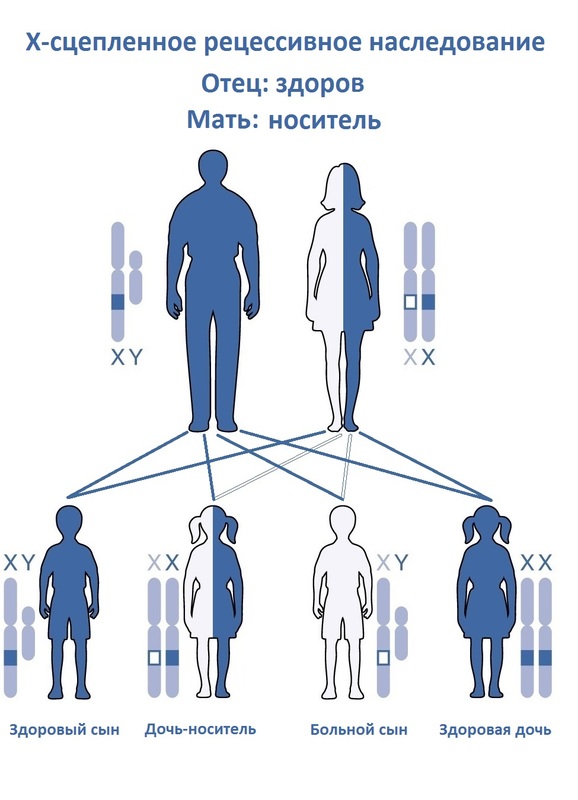
Синдром Хантера — это одна из форм мукополисахаридоза, генетическое заболевание, возникающее в результате дефицита ряда ферментов, расщепляющих продукты обмена. Этот дефицит приводит к накоплению белково‑углеводных комплексов и жиров в клетках. В результате чего происходит «самоотравление» организма и поражаются органы.
В России редким, или орфанным, считается заболевание, которое встречается у 1 человека из 10 тысяч или реже. Именно из-за редкости такие болезни сложно диагностировать и лечить. Тот синдром, что у Паши, встречается у одного ребенка на 132 000 родившихся младенцев.
Сейчас Снежана помогает больным мукополисахаридозом как президент МБОО «Хантер-синдром»: рассказывает, как получить лекарство, через благотворительные фонды находит памперсы, инвалидные коляски и ортопедическую обувь.
Право выбора
Большинство редких болезней хронические. То есть неизлечимые. Почти все приводят к инвалидизации и смерти. Орфанные препараты, если терапия от конкретного недуга все-таки существует, как правило, не излечивают болезнь полностью, а лишь снимают тяжесть симптомов и приниматься должны на протяжении всей жизни.
«С первой частью я согласилась, а вот кто такой Хантер, мне тогда было неизвестно»
Лечение двадцати четырех самых тяжелых и дорогих орфанных заболеваний финансируется государством. Если диагноз входит в знаменитый список «12 нозологий», то лекарство закупают за счет федерального бюджета, если в «Перечень 24» — за препараты платят регионы. В остальных случаях пациенты получают терапию как жизненно необходимую или по инвалидности.
Известно, что крайне важно начать лечение от мукополисахаридоза как можно раньше. Павлик получил первые свои медикаменты только в восемь. Сейчас ему девятнадцать, с восемнадцати он не говорит, хотя понимает человеческую речь.
«Конечно, мне часто пишут мамы, — признается Снежана: „Мы видели вашего ребенка в интернете. Он не разговаривает и ходит в памперсах. Зачем нужна такая жизнь?“. Но у меня короткий ответ. Что вы выберете: двенадцать лет ходить на кладбище или двенадцать лет целовать сына?».
За двенадцать лет терапии Павел не пропустил ни одного приема Элапразы. Хоть препарат и стоит около миллиона (!) рублей в неделю. Благо в Москве проблем с лечением нет.
Вопрос денег
У сына Натальи Буртаевой мукополисахаридоз IV типа, заболевание схожее, но ни в списке «12 нозологий», ни в «Перечне 24» его нет.
Даниилу уже шестнадцать. Его рост 97 сантиметров. Ходит с трудом: ноги и руки деформированы, внутренние органы тоже деформированы. С пятого класса Даня учится на дому. Нервная система при этом диагнозе не страдает, дети с ним отлично осваивают школьную программу. Но поврежденной оказывается опорно-двигательная система, страдает слух.
Препараты специальной ферментной заместительной терапии, положенной в таких случаях, существуют, но первое время были не зарегистрированы в России. Единственное лекарство, одобренное американским надзорным фармакологическим ведомством (FDA) в 2014 году, называется элосульфаза альфа — торговое название Вимизим (Vimizim).
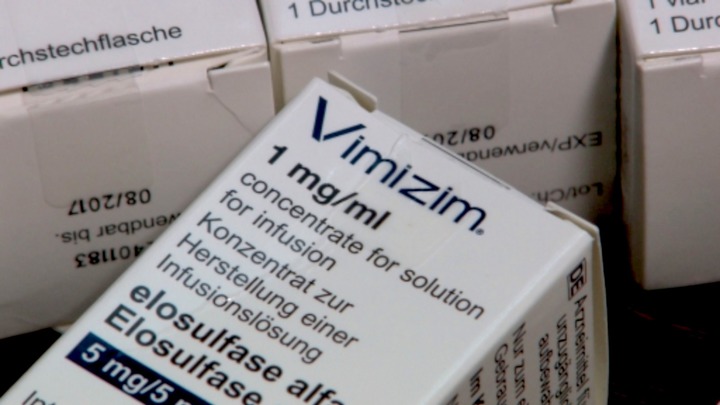
Без Вимизима больные мукополисахаридозом IV типа умирают к двадцати годам. Когда новости о его разработке только появились в специализированных СМИ, Наталья было обрадовалась. Но стоимость его годового курса превышает 60 миллионов рублей.
Минздрав Ульяновской области ожидаемо отказался приобретать препарат. Наталья подала в суд и выиграла: первый в 2016 году, второй, когда министерство подало апелляцию, — в 2017. Ни письма в Росздравнадзор и Путину, ни сюжет на «Первом канале» не помогли: Даниил до сих пор не получает лечения.
«Мы не прячемся, — твердо сообщает Наталья корреспонденту. — Даня часто гуляет на коляске. Мы с ним ездили на море, а сегодня вообще особенный день: последний звонок. Он выйдет на сцену и получит аттестат зрелости.
Добавляет: «Он у меня обычно говорит, когда я переживаю, что люди его не примут: „Мам, ты, это, не переживай. Я привык, что на меня так смотрят“».
Более того, после школы Даниил планирует учиться дальше, признается мать, вздыхая: «Не знаю, сколько он будет жить». Врачи точно ответить на этот вопрос тоже не в силах, но очевидно, что без терапии Даня проживет недолго.
«Сделано в России»
Почему орфанные препараты стоят так дорого? Фармакологические компании разрабатывают их ради прибыли. Исследования нового лекарства стоят немало, а весьма небольшой тираж препарата призван не только возместить потраченные на него миллионы, но и «вернуться» с процентами.
Если в производстве инсулина в России заинтересованы миллионы, то «целевая аудитория» орфанных препаратов даже на такую большую страну, как наша, нередко не превышает пары десятков пациентов.
Во многих странах предусмотрена государственная поддержка исследователей, разработчиков и производителей орфанных препаратов: льготы, гранты и так далее. Но в России таких программ нет.
«Мам, ты, это, не переживай. Я привык, что на меня так смотрят»
Для того чтобы снизить издержки, власти предпочитают не субсидировать или договариваться с производителем, а искать оригинальным препаратам дешевые аналоги, дженерики. К сожалению, как убеждены некоторые эксперты, они нередко отличаются по эффективности.
Насте Катасоновой двадцать один, и она не знает, сколько раз лежала в больнице: до восьми лет — раз в год, до четырнадцати — по два раза, потом — по три, сейчас — госпитализироваться приходится чуть ли не каждый месяц.
Говорит, что в больнице ей спокойно: за твоим состоянием следят специалисты. Впрочем, и тут есть поводы для волнений. У Насти муковисцидоз, редкая болезнь легких, и недавно оригинальные антибиотики заменили отечественным дженериком.
Внешне люди с редкими заболеваниями часто не отличаются от здоровых. Однажды в школе кто-то из родителей одноклассников сказал, что девочке родственники купили инвалидность, дабы та не ходила на уроки. Одноклассники в ответ на слух объявили несчастной бойкот. Теперь она о муковисцидозе говорит открыто, чтобы не возникало кривотолков.
В ноябре Насте двадцать два. Между больницами и процедурами она профессионально фотографирует, ведет личный блог, помогает с соцсетями благотворительным организациям, занимается веганским магазином.
От побочных эффектов нового дженерика — у оригинального препарата их не было — состояние Анастасии ухудшилось. «Но не пить их нельзя, иначе — захлебнешься в мокроте», — объясняет пациентка.
«Каждое утро для меня — испытание, — рассказывает она. — Мокрота за ночь отлеживается в легких. Встаю. Но не так красиво, как в фильмах, а с кашлем. За день получается стакан густой зеленой жижи. Таблетки, ингаляция, если надо на работу, приходится просыпаться за два часа до выхода». Днем опять ингаляции. Перед сном питание: через гастростому. Это специальную трубка, которая устанавливается в отверстие на животе и ведет прямо в желудок.
Екатерина Захарова, руководитель лаборатории наследственных болезней обмена веществ, председатель экспертного совета по редким болезням Всероссийского общества орфанных заболеваний, считает, что нужен активный диалог с разработчиками и производителями орфанных препаратов: «Необходимо выяснять, какие лекарства фармкомпании могут предложить бесплатно, какую минимальную стоимость могут установить. Кроме того, важно понять, какие препараты Россия может производить самостоятельно, чтобы обеспечить своих граждан: возможно, в рамках госзадания будет дешевле создать российское лекарство для небольшого числа пациентов».
Преступная забывчивость
Впрочем, не все орфанные препараты стоят миллионы. Вот только получить даже относительно недорогое лекарство нуждающимся в нем порой оказывается совсем не просто.
У Виктории Рыжковой редкое генетическое заболевание. Синдром Вильсона-Коновалова, или гепатоцеребральная дистрофия.
Медь при этой болезни не выводится из организма, а проблемы с нарушением ее обмена ведут к накоплению элемента в нервной системе, почечной, печеночной тканях и роговице, что оборачивается токсическим повреждением указанных органов.
Орфанные болезни
Опубликовано в журнале:
« Практика педиатра » № 4 Ноябрь-декабрь, 2019
Материал подготовлен совместно с:
Екатерина Захарова, д.м.н., профессор кафедры медицинской генетики ФГБОУ ДПО «Российская медицинская академия непрерывного профессионального образования» МЗ РФ; зав. лабораторией наследственных болезней обмена веществ ФГБНУ «Медико-генетический научный центр им. акад. Н.П. Бочкова», председатель экспертного совета Всероссийского общества орфанных заболеваний;
Елена Красильникова, аналитик, руководитель проектного офиса «Редкие (орфанные) болезни» ФГБНУ «Национальный научно-исследовательский институт общественного здоровья им. Н.А. Семашко»
— В последнее время орфанные заболевания обсуждаются все чаще как в специализированных кругах, так и широкой общественностью. С чем это связано?
Тема редких заболеваний относительно «молодая». Диагностика и лечение редких заболеваний стали доступны в конце XX в. благодаря развитию генетического тестирования и биотехнологий.
Впервые с редкими нозологиями система здравоохранения РФ столкнулась после анализа заявок на лекарственные препараты в рамках программы дополнительного лекарственного обеспечения в 2006-2007 гг. Было выявлено 7 нозологий, требующих применения лекарственных препаратов с высокой стоимостью, в том числе 4 редких заболевания (болезнь Гоше, муковисцидоз, гипофизарный нанизм, гемофилия). Тогда еще система здравоохранения не выделяла их как орфанные, потому что в России не было специального законодательства в отношении редких болезней.
8 лет назад, в 2011 г., в Федеральном законе № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» впервые появилось понятие «редкое (орфанное) заболевание» и начала формироваться система учета пациентов (Федеральный регистр), страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, приводящими к сокращению продолжительности жизни или к инвалидности. Одновременно с этим обязательства по лекарственному обеспечению граждан, страдающих такими заболеваниями, были возложены на субъекты РФ.
Конечно, это пока возможно для очень небольшого числа (около 250) из известных редких болезней (их описано более 7000, и с каждым годом появляются данные о все новых заболеваниях).
Ну и поскольку таких пациентов немного, лечение для них довольно дорогостоящее, в некоторых регионах РФ треть всего бюджета на лекарственное обеспечение тратится именно на орфанных пациентов.
— Как быстро ставится такой диагноз? Какие проблемы с диагностикой существуют в РФ?
Во всем мире постановка редкого диагноза занимает до 7 лет. Почему так сложно его установить? Врач, который наблюдает пациента, педиатр или терапевт, сначала подозревает какие-то более распространенные заболевания и только методом исключения постепенно выходит на редкое. В большинстве случаев точный диагноз возможно установить только благодаря труду и упорству врача, лаборатории, самого пациента и его семьи.
Есть разные диагнозы и разный маршрут пациентов. Иногда путешествие за своим диагнозом занимает годы, а иногда врач, уже знакомый с данной патологией, может почти безошибочно поставить диагноз на первом приеме, при первом взгляде на пациента. Например, при мукополисахаридозах (МПС) есть внешние особенности, которые если знаешь, ни с чем не перепутаешь. А врачи-ортопеды, которые занимаются редкой патологией, глядя на рентгеновские снимки, могут высказать предположение о синдроме Моркио (МПС IVA типа), так как при нем наблюдаются особенности строения многих костей, задержка роста, деформация скелета.
Но попасть к нужному специалисту не всегда удается, а многие врачи, конечно, недостаточно знают эту патологию.
— Многие редкие заболевания проявляются у ребенка с ранних лет. Можно ли сказать, что при оказании надлежащей помощи многие люди, страдающие орфанными заболеваниями, смогут прожить значительно дольше? Если да, то каких диагнозов это касается в первую очередь? Какие патологии проявляются раньше всего?
Во многих случаях ранняя диагностика и начало лечения помогут избежать тяжелых осложнений, предотвратить раннюю инвалидизацию и летальный исход. Не говоря уже об улучшении качества жизни детей и членов их семей. Если вовремя поставить диагноз, такой как нарушение окисления жирных кислот или дефекты обмена органических кислот и аминокислот, то вовремя начатая диетотерапия позволяет спасти жизнь ребенка, и пациенты с этими болезнями могут жить практически столько же, сколько обычные люди, при условии соблюдения диеты.
Многие из этих заболеваний включены в программы массового скрининга новорожденных во многих странах мира. В РФ скрининг пока проводится только на 5 болезней, но вопрос о расширении скрининговых программ уже обсуждается и в Министерстве здравоохранения, и в Правительстве РФ.
— Расскажите отдельно про такие заболевания, как МПС и фенилкетонурия. Как типы МПС различаются между собой? Какое место эти патологии занимают в структуре детской заболеваемости?
Поскольку эти болезни редкие, конечно, они занимают не очень существенное место в структуре детской заболеваемости.
— Какие сегодня существуют актуальные подходы к диагностике и лечению МПС и ФКУ? Расскажите про доступность медицинской помощи и лекарственного обеспечения пациентам с редкими заболеваниями в РФ. Насколько выстроена помощь больным в Москве и какова ситуация в регионах? Согласно статистике в каких регионах наблюдается самая тяжелая ситуация с орфанными пациентами? С чем это связано?
Лечение ФКУ заключается в соблюдении специальной диеты. По закону при амбулаторном лечении этого заболевания следующие лекарственные средства и изделия медицинского назначения отпускаются по рецептам врачей бесплатно: безбелковые продукты питания, белковые гидролизаты, ферменты, психостимуляторы, витамины, биостимуляторы.
Необходимость обеспечения льготным лекарственным препаратом «редкого» пациента привела к беспрецедентному росту расходов на эти цели в Приморском и Камчатском краях, Волгоградской, Ивановской и Тамбовской областях, Республиках Калмыкия и Чувашия.
Основное финансовое бремя сформировали 10 жизнеугрожающих редких нозологий: пароксизмальная ночная гемоглобинурия (болезнь Маркиафавы-Микели), идиопатическая тромбоцитопеническая пурпура (синдром Эванса), МПС (типы I, II и VI), гемолитико-уремический синдром (ГУС), легочная (артериальная) гипертензия (идиопатическая) (первичная), нарушения обмена ароматических аминокислот (классическая ФКУ, другие виды гиперфенилаланинемии), юношеский артрит с системным началом (ЮАС), другие сфинголипидозы: болезнь Фабри (Фабри-Андерсона), болезнь Ниманна-Пика. Расходы на льготное лекарственное обеспечение пациентов с этими нозологиями в 2017 г. составили 14,8 млрд руб., или 86% совокупного бюджета субъектов РФ на эти цели. При этом дефицит финансирования лекарственного обеспечения данной группы нозологий в среднем составил 46%.
В 2019 г. перевод на федеральный уровень лекарственного обеспечения пяти жизнеугрожающих редких заболеваний с преимущественно детской когортой пациентов (ГУС, ЮАС, МПС I, II, VI типов) сократил соответствующий регистр на 12% и предоставил возможность лечения за счет федерального бюджета каждому пятому ребенку, включенному в этот регистр.
Принятое федеральной властью решение позволит сократить дефицит финансирования лекарственного обеспечения в ряде наиболее неблагополучных регионов: в Республиках Ингушетия и Дагестан, Брянской области, Удмуртской и Кабардино-Балкарской Республиках и т.д. Однако в некоторых регионах из-за специфики эпидемиологии жизнеугрожающих редких заболеваний и отсутствия (или незначительного количества) пациентов с нозологиями, лекарственная поддержка при которых обеспечивается федеральным бюджетом, ситуация, к сожалению, останется неизменной, например в Смоленской области, Карачаево-Черкесской Республике.
В среднем уровень обеспеченности жизненно необходимыми лекарственными препаратами у пациентов с жизнеугрожающими редкими заболеваниями в 2017-2018 гг. составил 54%. Регионами с наименьшей обеспеченностью стали: Забайкальский край, Республики Татарстан, Ингушетия, Удмуртская и Кабардино-Балкарская. Здесь лекарственную помощь получают меньше 1/3 нуждающихся «редких» пациентов.
Ситуация с лекарственным обеспечением таких пациентов в рамках федеральной программы гораздо благополучнее.
— Что поможет оптимизировать расходы государства и регионов?
Федерализация, регулирование цен государством могли бы помочь в данном вопросе. Допустим, регион закупает орфанные препараты на одного или двух пациентов, а в стране, например, с этим диагнозом живут 100 человек. Сконцентрировав закупки «в одних руках», федеральное министерство может вести переговоры с представителем производителя о снижении цены или использовании инновационных подходов к финансированию, таких как «разделение рисков», «закрытый контракт» и т.д.
В 2019 г. практически вдвое была снижена цена на ряд препаратов, закупаемых ранее в рамках региональной программы, благодаря тому, что лекарственное обеспечение 5 нозологий перевели на федеральный уровень.
Также если пациентов начинают своевременно лечить и не доводят их до более тяжелых состояний, то будут нужны меньшие дозировки лекарственных средств. Когда система четко работает, она требует меньше затрат.
— Расскажите подробнее про организацию медицинской помощи пациентам с редкими заболеваниями, не включенными в льготные программы федерального и регионального уровней, в том числе про обеспечение незарегистрированными лекарственными препаратами. Различается ли медикаментозная терапия у детей и взрослых? Есть ли препараты, находящиеся в постоянном дефиците?
С одной стороны, в законодательстве Российской Федерации предусмотрена возможность назначения, ввоза и закупки незарегистрированных препаратов по жизненным показаниям. С другой стороны, пациентам крайне сложно этого добиться: практически в 100% случаев это происходит только через суд, поскольку нет четкого разъяснения по следующим вопросам: из каких средств может оплачиваться закупка; как защищены врач и лечебное учреждение при неблагоприятном исходе терапии, если препарат не упомянут в клинических рекомендациях и стандартах оказания медицинской помощи?
Если незарегистрированный препарат назначается федеральным консилиумом, то почему за его применение юридическую ответственность (перед пациентом) несет лечащий врач из региона, который может и не разделять мнение консилиума или совершенно ничего не знать про это редкое заболевание? И если препарат назначает федеральное учреждение, то почему на это должны тратиться средства исключительно регионального бюджета?
Есть и другая проблема: если регион согласен закупить препарат, но у пациента нет инвалидности (например, это больной, у которого еще нет симптомов болезни), то юридически невозможно такую закупку обосновать.
Таким образом, вопросов очень много.
Этих пациентов не так много, но лекарственные препараты для них очень дорогие. Вот некоторые из этих нозологий: нейрональный цероидный липофусциноз II типа, нарушения синтеза желчных кислот, нарушения цикла мочевины. Они не входят в перечень высокозатратных нозологий или орфанный перечень, который утверждается Правительством РФ, и препараты для них не зарегистрированы.
— Можно ли сказать, что сегодня российская система здравоохранения адаптирована к большому количеству больных редкими заболеваниями?
Нет ни одной системы здравоохранения в мире, которая раз и навсегда адаптирована к большому количеству больных редкими заболеваниями. Появляются новые методы лечения, каждый из которых дороже предыдущего. Система всякий раз вынуждена адаптироваться. Сейчас всё настолько быстро меняется с точки зрения технологий, что каждые 2-3 года систему нужно совершенствовать.
Говорить об этом. Заниматься образованием врачей, постоянно повышать их квалификацию. Нужно донести до обычного врача, что в том случае, когда он не может диагностировать болезнь, есть большая вероятность развития редкого заболевания.
— Разумно ли говорить о профилактике редких заболеваний, можно ли «застраховать» от этого своего будущего ребенка?
О профилактике редких заболеваний можно говорить в двух случаях: если в семье уже есть ребенок с патологией либо известно, что родители являются носителями болезни. Таким семьям необходима консультация врача-генетика, который может предложить им пренатальную, преимплантационную диагностику, использование донорских клеток, что позволит родить здорового ребенка.
Орфанные заболевания. Редкие и опасные

Обсуждаемой новостью стало, что часть средств, вырученных от увеличения ставки НДФЛ, планируется направить в целевой фонд на лечение детей с орфанными заболеваниями. Подробнее об орфанных заболеваниях и как они изучаются в России в нашем материале.
Что такое орфанные заболевания?
Редкость заболевания, с одной стороны, плюс, так как немногие люди сталкиваются с ними, но для носителей заболевания – существенный минус. Недостаточная изученность болезни затрудняет постановку диагноза, а в следствии, и оказание своевременного лечения.
Минздрав России 27 февраля 2020 года опубликовал перечень орфанных заболеваний, куда входят 258 болезней, включая различные новообразования, заболевания кровообращения, эндокринные нарушения, врождённые аномалии, психические расстройства и другие.
Лечение орфанных заболеваний дорогостоящее. Например, для лечения детей со спинальной мышечной атрофией применяют препарат стоимостью около восьми миллионов рублей за одну инъекцию.
В России существуют программы по лечению орфанных заболеваний на региональном и федеральном уровнях, но многие родители вынуждены обращаться к помощи благотворительных фондов, самостоятельному поиску средств на лечение детей через Интернет. В такой ситуации необходима системная поддержка на государственном уровне.
Как изучаются орфанные заболевания?
По информации Минобрнауки, в России существует всего несколько научных центров и лабораторий, где изучаются орфанные заболевания и генные патологии, в том числе Лаборатория «Молекулярная медицина и генетика человека» СВФУ, Лаборатория терапии орфанных заболеваний МФТИ, Медико-генетический научный центр имени академика Н.П. Бочкова. Эти лаборатории занимаются изучением причин и механизмов развития орфанных и мультифакториальных заболеваний, разрабатывают способы диагностики и профилактики этих заболеваний.
Особое место в работе лаборатории «Молекулярная медицина и генетика человека» СВФУ занимает исследование генетических заболеваний, характерных для представителей якутского этноса, в связи с чем она работает в тесном сотрудничестве с Национальным центром медицины Республики Саха (Якутия). Изучаются: SOPH-синдром, приводящий к низкорослости с атрофией зрительных нервов, 3-М синдром, так называемый «якутский синдром низкорослости», врожденная глухота первого А-типа, наследственная метгемоглобинемия и синдром мукополисахаридоз-плюс и другие заболевания. Все разработки СВФУ – новые методы диагностики и лечения – сразу внедряются в практическое здравоохранение.
В лаборатории терапии орфанных заболеваний МФТИ при совместной работе с Национальным медицинским исследовательским центром эндокринологии Минздрава РФ специалисты работают над созданием терапии для врожденной дисфункции коры надпочечников – группы заболеваний, обусловленных нарушением стероидогенеза в результате дефицита одного из ферментов, участвующих в синтезе кортизола. Предлагаемая лабораторией стратегия терапии основывается на интеграции нормальной копии гена в геном стволовых клеток коры надпочечников для обеспечения пожизненного терапевтического эффекта, что станет альтернативой применяющейся гормональной терапии. Кроме того, в лаборатории геномной инженерии МФТИ по заказу компании «Артема» ведется разработка генной терапии колбочковой дистрофии сетчатки глаза с измененным палочковым ответом. В случае успешного тестирования, терапия позволит остановить потерю зрения у людей с генной мутацией, а также поможет восстановить зрение уже ослепшим из-за наследственной патологии пациентам.
Сотрудники Медико-генетического научного центра имени академика Н.П. Бочкова разрабатывают уникальные методы эффективной диагностики наследственных болезней, а также новые методы лечения, лекарственные средства, способы диагностики и индивидуализации терапии онкологических заболеваний. Для каждого конкретного пациента разрабатывается новый, уникальный метод функционального анализа. В 2019 году специалисты МГНЦ осуществили консультативный прием 11 тысяч пациентов и провели свыше 13 тысяч генетических исследований в рамках госзадания. Центр является первым в Европе по количеству диагностируемых наследственных заболеваний и единственной организацией в России, выполняющей высокотехнологичные генетические тесты на бюджетной основе в таком объеме.
«Уровень развития медицинской генетики в нашей стране соответствует общемировому. В России сегодня есть возможности применения всех современных подходов диагностики и профилактики наследственных болезней. Развиваются полногеномные исследования, анализ хромосом, подходы профилактики наследственной и врожденной патологии. При этом технологии, которыми владеют врачи в Российской Федерации, не отличаются от технологий, применяющихся в развитых странах. Сегодня у нас меняется парадигма: мы говорим уже не о симптоматическом, а о патогенетическом лечении, о развитии таких современных технологий, как генотерапия. Мы подошли очень близко к тому, чтобы получить лекарственные препараты для лечения ряда наследственных заболеваний. Совместная работа с международным консорциумом Orphanet позволит сделать дальнейшие шаги в этом направлении», – сообщил Сергей Куцев, директор ФГБНУ «МГНЦ», главный внештатный специалист по медицинской генетике Минздрава России.



