Что такое периодическая болезнь
Периодическая болезнь, или средиземноморская семейная лихорадка, — наследственное заболевание. Для нее характерны кратковременные острые и самопроизвольно исчезающие подъемы температуры тела, периоды полисерозита, возобновляющегося с нерегулярными интервалами, а также амилоидоз, который в отсутствие лечения приводит к тяжелой почечной недостаточности. Тип наследования — аутосомно-рецессивный. Болезнь встречается главным образом у выходцев из Средиземноморья — евреев-сефардов, турок, apмян и арабов, среди которых частота носительство патологического гена достигает 20 %.
Среди греков, испанцев и итальянцев она несколько ниже. У евреев-ашкенази, немцев и англосаксов периодическая болезнь наблюдается редко, а среди других этнических групп встречается лишь спорадически. В 63-68 % случаев первые клинические проявления возникают до 5-летнего возраста, в 90% случаев — до 20-летнего. Самый ранний возраст начала заболевания — 6 мес. Типичные острые периоды продолжаются 1-4 сут, сопровождаясь лихорадкой и симптомами перитонита (90%), артрит, или артралгии (85%) и плеврита (20%). Перикард и влагалищная оболочка яичка поражаются редко.
Ген периодической болезни был картирован в небольшом сегменте короткого плеча хромосомы 16 (р13,3) между генами поликистоза почек и синдрома Рубинстайна-Тейби. Затем двум исследовательским группам (международной и французской) удалось выделить и клонировать этот ген, получивший название MEFV. Он принадлежит к семейству генов RoRet, насчитывает примерно 10 тыс. пар оснований и содержит 10 экзонов. Его транскрипт (3,7 тыс. пар нуклеотидов) кодирует состоящий из 761 аминокислот белок пирин (от греч. «огонь»), или маренострин (от лат. «наше море»), который экспрессируется в миелоидных клетках. Обнаружено более 29 мутаций гена MEFV, большинство которых локализуется в последнем экзоне. Все ли они ассоциированы с периодической болезнью, неясно.
Почти у 70 % больных средиземноморского происхождения встречаются те или иные из пяти наиболее частых мутаций (M694V, V726A, M694I, M6801 и E148Q). Анализ гаплотипов и мутаций среди носителей дефектов показывает, что, происходя от общего предка, они в течение столетий разделились. Наиболее частая миссенс-мутация — метионин-694-валин (M694V) — встречается в 30-67% случаев и ассоциируется с тяжелым течением заболевания и высокой частотой амилоидоза. На 2-м месте находится мутация валин-726-аланин (7-35%), ассоциирующаяся с более легким течениям болезни и меньшей частотой амилоидоза. Таким образом, фенотипические различия болезни могут определяться разными мутациями. Клонирование идентификация гена периодической болезни сделали возможной ее диагностику даже там, где она встречается редко и мало знакома врачам. Генетическии скрининг (с помощью ПЦР) теперь проводят в ряде лабораторий.
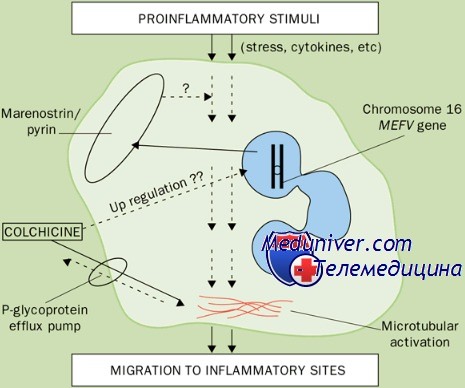
Однако в таких лабораториях обычно определяют только 5-10 наиболее частых мутаций, пропуская более редкие. Поэтому диагностика периодической болезни все еще основывается на клинической картине, а генетические исследования проводят лишь для подтверждения диагноза. Патогенез обострений болезни остается мало изученным, хотя имеются сообщения о некоторых имунных нарушениях. Особый интерес представляют данные о недостаточности ингибитора (инактивирующего фермента) С5а. Компонент комплемента С5а — анафилатоксин, обладающий высокой хемотаксической активностью. В норме небольшие количества С5а, попадающие в серозные полости, нейтрализуются инактивирующим ферментом еще до того, как вызовут воспаление. Одна из гипотез патогенеза периодической болезни заключается в том, что недостаточность ингибитора С5а (вследствие аномальности пирина) обусловливает накопление его в серозных полостях, что и лежит в основе острых приступов заболевания.
Дальнейшее изучение функции пирина должно пролить свет на его взаимодействие с другими белками, принимающими участие в воспалительной реакции.
Приступы периодической болезни можно предотвратить приемом колхицина в дозе 0,02—0,03 мг/кг (максимум 2 мг) в сутки. Суточную дозу принимают за 1 или 2 раза. Лечение колхицином снижает не только частоту приступов, но и риск амилоидоза, а также уменьшает степень уже имеющегося амилоидоза. Прием колхицина во время беременности, по-видимому, не опасен ни для матери, ни для плода.
В последнее время обнаружено по крайней мере три других синдрома периодической лихорадки, которые следует отличать от периодической болезни. Гипер-IgD периодический лихорадочный синдром (HIDS) — аутосомно-рецессивное заболевание, встречающееся преимущественно в семьях европейского происхождения (голландцев, французов). В его основе лежат мутации гена мевалонаткиназы, локализованного на хромосоме 12 (участок q24). Приступы лихорадки продолжаются более 14 сут и сопровождаются увеличением шейных лимфатических узлов, болью в животе, сыпью, артралгией/артритом и иногда спленомегалией. В крови возрастает уровень белков острой фазы воспаления и IgD (> 100 ЕД/мл).
Специфические методы лечения неизвестны, хотя иногда помогают глюкокортикоиды. Периодический синдром, ассоциированный с рецептором ФНО (TRAPS), ранее носил другие названия: семейная ирландская лихорадка, семейная периодическая лихорадка и аутосомно-доминантная повторяющаяся лихорадка. Описано лишь несколько случаев этого синдрома в семьях ирландского и шотландского происхождения. В основе лежат мутации гена, расположенного на хромосоме 12 (участок р13) и кодирующего рецептор ФНО 1-го типа. У больных возникают кратковременные (4-6 сут) подъемы температуры, сопровождающиеся болью мышц живота. Могут иметь место сыпь, конъюнктивит и односторонний периорбитальный отек.
Иногда возрастает содержание белков острой фазы воспаления, но наиболее специфичный признак — низкий уровень растворимого рецептора ФНО 1-го типа в сыворотке крови. Для лечения применяют глюкокортикоиды. Показана терапевтическая эффективность этанерцепта (рекомбинантного рецептора ФНО). Недавно описано заболевание, названное синдромом периодической лихорадки с афтозным стоматитом, фарингитом и аденитом (PFAPA). У некоторых больных наблюдалась и артралгия. В большинстве случаев симптомы исчезали уже после однократного приема преднизона (1-2 мг/кг). Обычно этот синдром развивается до 5-летнего возраста и через 4-8 лет самопроизвольно исчезает. Неясно, имеет ли он инфекционную природу или связан с иммуногенетическими нарушениями.
Периодическая болезнь
Периодическая болезнь – генетическая патология, характеризующая нарушением регуляции воспалительных процессов, особенно в области серозных (брюшины, плевры) и синовиальных оболочек. Проявления этого заболевания различны, чаще всего регистрируются боли в животе (картина острого перитонита), нарушения со стороны плевральной полости, приступы лихорадки, болезненность и отечность суставов. Диагностика производится на основании клинической картины, изучения наследственного анамнеза и молекулярно-генетических анализов, вспомогательную роль играет определение национальности больного. Лечение периодической болезни только симптоматическое и поддерживающее, специфической терапии на сегодняшний момент не существует.
Общие сведения
Периодическая болезнь (средиземноморская семейная лихорадка) – наследственное заболевание, причиной которого являются нарушения в регуляции воспалительного и иммунного ответа на уровне гранулоцитов. Впервые данная патология была описана в 1948-м году американским врачом Райманном, который из-за повторяющихся тяжелых приступов дал ей название «периодическая болезнь». С первых лет изучения была выявлена главная особенность этой патологии – она возникает только у уроженцев средиземноморского региона и Малой Азии, главным образом у армян, арабов, греков, испанцев, итальянцев, евреев-сефардов и турок. У представителей иных национальностей отмечены лишь спорадические и статистические незначимые случаи периодической болезни. Поэтому фактор национальности больного и его предков играет немаловажную роль в диагностике данного состояния.
Встречаемость периодической болезни у различных этносов средиземноморского региона отличается, она наиболее высока у турок, арабов и армян, несколько ниже у евреев-сефардов, еще реже это заболевание встречается у греков, итальянцев и испанцев. По некоторым данным, носительство патологического гена в определенных регионах затрагивает 20% населения, а заболеваемость составляет 1:1000-2500. Периодическая болезнь наследуется по аутосомно-рецессивному механизму и с одинаковой частотой поражает как мальчиков, так и девочек.
Причины
Долгое время этиология и патогенез периодической болезни оставались неизвестными, лишь достижения современной генетики позволили больше узнать об этом заболевании. Наиболее часто причиной данной патологии являются мутации гена MEFV, расположенного на 16-й хромосоме. Ген кодирует белок под названием маренострин (другое название – пирин), который выполняет функции одного из центральных регуляторов воспалительной реакции и первичного иммунного ответа. Маренострин тормозит дегрануляцию нейтрофилов и угнетает их адгезивные свойства, тем самым ослабляя и ингибируя чрезмерную реакцию иммунной системы. При периодической болезни миссенс-мутации гена MEFV приводят к изменению структуры маренострина, тем самым нарушая его функции. Это снижает порог дегрануляции нейтрофилов, что облегчает развитие острых воспалительных реакций и формирует клиническую картину периодической болезни.
Кроме того, дефекты маренострина приводят к каскадным патологическим реакциям в иммунной системе и организме в целом. Значительно уменьшается активность ингибитора одного из компонентов системы комплемента – С5а. Последний постепенно накапливается в серозных оболочках, а при достижении высоких концентраций провоцирует бурную воспалительную реакцию. Это обстоятельство объясняет определенные свойства периодической болезни – преимущественное поражение серозных оболочек, а также сезонность заболевания (для накопления достаточных концентраций С5а необходимо несколько месяцев). В некоторых случаях для периодической болезни характерно также раннее развитие амилоидоза, однако его патогенез остается неясным.
Все вышеперечисленные процессы возникают при наличии у человека двух аллелей дефектного гена MEFV, то есть у гомозигот, так как периодическая болезнь является аутосомно-рецессивным заболеванием. Существует теория, согласно которой гетерозиготы из-за снижения ингибирования адгезивных свойств гранулоцитов обладают повышенной резистентностью к бактериальным инфекциям. Отчасти это может объяснить столь высокую встречаемость патологической формы гена и его носительства среди этносов средиземноморского региона. Кроме того, существуют указания, что некоторые формы периодической болезни обусловлены дефектом генов на 19-й хромосоме, однако точно идентифицировать их пока не удалось.
Симптомы периодической болезни
Клиническая картина периодической болезни отличается большим разнообразием, однако причины этого пока достоверно неизвестны – предполагается взаимосвязь между отдельными типами мутаций и формами заболевания. Удалось выяснить, что, например, амилоидоз, в среднем поражающий 30-35% больных, намного чаще возникает у арабов и турок, нежели у армян. Постоянным симптомом периодической болезни (наблюдается в 99% случаев) является выраженная лихорадка, которая не купируется традиционными жаропонижающими средствами и антибиотиками. В зависимости от клинической формы заболевания повышение температуры тела может сочетаться с другими проявлениями. На сегодняшний день выделяют четыре основные клинические формы периодической болезни: абдоминальную, торакальную, суставную и псевдомалярийную.
Абдоминальная форма периодической болезни характеризуется типичной картиной «острого живота» при перитоните, включает в себя резкое повышение температуры тела до 40-41 градуса, опоясывающие боли, ригидность мышц брюшной стенки, тошноту и рвоту. Такие проявления сохраняются на протяжении нескольких суток, после чего постепенно стихают. За это время более чем у половины больных периодической болезнью ошибочно диагностируется гнойный перитонит (на самом деле при данной патологии развивается асептическое воспаление брюшины), аппендицит, прободная язва желудка, производятся ненужные хирургические операции. Торакальная форма периодической болезни создает картину выпотного плеврита, который может иметь одно- или двухсторонний характер. Это приводит к болям в грудной клетке, затрудненному дыханию, одышке и другим типичным проявлениям гнойного или экссудативного плеврита, что также нередко становится причиной постановки ошибочного диагноза. Проявления торакальной формы периодической болезни постепенно стихают на протяжении 7-10 дней.
Суставная форма периодической болезни характеризуется развитием отеков и болезненности нескольких (реже – одного) суставов, резким покраснением кожи на пораженной области. Симптомы сохраняются на протяжении 2-4 недель, артралгия может наблюдаться в течение нескольких месяцев. При этом каких-либо постоянных нарушений в суставах (ограничение подвижности, контрактуры) при периодической болезни не возникает. Псевдомалярийная форма заболевания характеризуется приступами сильной лихорадки длительностью 3-7 дней, после чего температура тела больного приходит в норму. Никаких проявлений со стороны других органов на начальных этапах развития патологии при этом не определяется.
По данным медицинской статистики, изолированные клинические формы (абдоминальная, торакальная и другие) имеют место примерно в 20% случаев периодической болезни. Намного чаще встречается сочетание нескольких клинических типов патологии (торакальной и суставной, лихорадки на фоне абдоминальных симптомов). При отсутствии лечения периодической болезни примерно у трети больных развивается амилоидоз почек, который приводит к хронической почечной недостаточности и уремии. В 20% случаев на фоне вышеперечисленных проявлений могут отмечаться дерматологические симптомы: папулезная сыпь, крапивница, рожеподобное воспаление. Редко при периодической болезни развиваются асептические менингиты и перикардиты, а также воспаление яичек (орхит).
Диагностика
В ряде случаев диагностика периодической болезни может быть сопряжена со значительными сложностями по причине выраженности, и, в то же время, неспецифичности ее проявлений. Эта особенность заболевания может стать причиной диагностических ошибок с далеко идущими последствиями – так, при картине «острого живота» больным часто производят ненужные операции, при асептических плевритах и менингитах назначают высокие дозы антибиотиков. В случае артралгии и постановки неправильного диагноза (например, ревматоидный артрит) больному периодической болезнью могут назначать сильнодействующие иммуносупрессивные средства. Поэтому при наличии таких симптомов у пациентов, являющихся уроженцами средиземноморского региона, обязательно следует учитывать возможность наличия этого генетического заболевания.
В процессе диагностики периодической болезни используют данные изучения наследственного анамнеза больных и молекулярно-генетических анализов. Как правило, наследственный анамнез у таких пациентов отягощен (крайне редко встречаются спорадические формы), подобные проявления выявляются у предков или родственников. Окончательно подтвердить или опровергнуть наличие периодической болезни может врач-генетик посредством генетического исследования. Существует распространенная методика поиска наиболее часто встречающихся при этом заболевании мутаций гена MEFV – M694V и V726A, которые обуславливают более 75% всех случаев данной патологии. Однако более редкие дефекты MEFV могут остаться незамеченными – для их определения применяют секвенирование всей последовательности гена.
Лечение периодической болезни
Прогноз и профилактика
Прогноз периодической болезни в значительной степени зависит от наличия или отсутствия амилоидоза. Если его нет, несмотря на тяжелые приступы заболевания, прогноз благоприятный, поскольку в межприступный период больные чувствуют себя удовлетворительно, продолжительность жизни практически не сокращается. В случае развития амилоидоза на фоне периодической болезни выживаемость пациентов резко снижается из-за поражения почек. Риск возникновения амилоидоза уменьшается при ранней диагностике средиземноморской семейной лихорадки и своевременно начатом лечении колхицином. Профилактика периодической болезни возможна только в рамках пренатальной диагностики, которая рекомендуется в тех случаях, когда у обоих родителей имеется подозрение на носительство дефектной формы гена MEFV.
Периодическая болезнь и почечный амилоидоз у детей
Периодическая болезнь (синонимы: семейная средиземноморская лихорадка, доброкачественный пароксизмальный перитонит, рецидивирующий полисерозит, еврейская болезнь, армянская болезнь) — наследственное аутосомно-рецессивное заболевание, распространенное сред
Периодическая болезнь (синонимы: семейная средиземноморская лихорадка, доброкачественный пароксизмальный перитонит, рецидивирующий полисерозит, еврейская болезнь, армянская болезнь) — наследственное аутосомно-рецессивное заболевание, распространенное среди представителей древних народов Средиземноморья. Наиболее часто периодическая болезнь (ПБ) встречается у евреев-сефардов, армян, арабов, греков, турков, народов Кавказа и т. д., отсюда другие названия заболевания. Встречаемость ПБ среди евреев-сефардов по разным данным составляет от 1:250 до 1:2000 (частота носительства мутантного гена от 1:16 до 1:8), среди армян — от 1:100 до 1:1000 (частота носительства — от 1:7 до 1:4).
Среди 15 детей с ПБ, наблюдавшихся в Российской детской клинической больнице (РДКБ) в течение последних лет, 8 были армянами, 4 — дагестанцами, 1 — грек, 1 — с чеченскими и еврейскими корнями, 1 — русский.
Этиология и патогенез
В основе ПБ лежит точечная мутация в гене белка пирина, расположенного в коротком плече 16-й хромосомы (16q) рядом с генами аутосомно-доминантного поликистоза почек и туберозного склероза. Пирин — белок первичных гранул нейтрофилов, активно участвующий в регуляции процессов воспаления. Считается, что пирин стимулирует выработку противовоспалительных медиаторов, позволяет контролировать хемотаксис, стабилизирует мембрану гранулоцитов. Нарушение структуры этого белка, имеющее место при ПБ, приводит к повышению выработки провоспалительных медиаторов в лейкоцитах, активации микротубулярного аппарата и спонтанной дегрануляции первичных гранул лейкоцитов, активации молекул адгезии и усиленному хемотаксису лейкоцитов, результатом чего является воспаление.
На сегодняшний день известно 8 типов мутаций в С-концевом участке гена пирина, при которых происходит точечная аминокислотная замена. Наиболее распространены три мутации, на долю которых приходится более 90% случаев ПБ: М680I (замена изолейцина на метионин), М694V (замена валина на метионин), V726А (замена аланина на валин). Все три мутации насчитывают 2000–2500 лет, поэтому их иногда называют «библейскими», поэтому имеют преимущественное распространение у представителей древних народов, населявших земли вокруг Средиземного моря. Мутация М680I встречается в основном у армян, М694V и V726А — у всех этнических групп.
ПБ протекает в виде приступов, основой которых является спонтанная или спровоцированная дегрануляция нейтрофилов с выбросом медиаторов и развитием асептического воспаления преимущественно на серозных и синовиальных оболочках. В периферической крови повышается количество нейтрофилов и острофазовых белков (СРБ — С-реактивный реактивный белок, SAA — сывороточный белок амилоида А и др.). Раздражение медиаторами воспаления рецепторов приводит к развитию болевого синдрома, а воздействие большого количества эндогенных пирогенов на центр терморегуляции — к развитию лихорадки.
Клиническая картина и течение
Клинически ПБ проявляется возникающими через определенные интервалы (дни — недели — месяцы) стереотипными приступами лихорадки. Лихорадке могут сопутствовать болевые синдромы, связанные с развитием неспецифического воспаления в серозных и синовиальных покровах. В зависимости от пенетрантности генов эти синдромы могут быть изолированными или сочетаться, но каждый из них сохраняет свой ритм. Любая атака сопровождается лейкоцитозом, увеличением СОЭ и других воспалительных белков, повышением a- и b-фракции глобулинов, снижением активности миелопероксидазы нейтрофилов. Вне приступа дети чувствуют себя хорошо, лабораторные показатели постепенно нормализуются.
Лихорадка — наиболее частый и постоянный симптом при ПБ, встречается в 96–100% случаев. Особенностью лихорадки при ПБ является то, что она «не контролируется» антибиотиками и антипиретическими средствами. Изолированная лихорадка при ПБ, как правило, приводит к диагностическим ошибкам и расценивается как проявление ОРВИ.
Вторым по частоте симптомом ПБ является абдоминальный болевой синдром (асептический перитонит), который встречается в 91% случаев, а изолированно — в 55%. Клинически асептический перитонит мало отличается от септического со всем характерным для последнего симптомокомплексом: температура до 40°, резчайшая абдоминалгия, тошнота, рвота, угнетение перистальтики кишечника. Через несколько дней перитонит стихает, перистальтика восстанавливается. Подобная клиника часто является причиной диагностических ошибок, и больных оперируют по поводу острого аппендицита, перитонита, холецистита, кишечной непроходимости и др. Среди наблюдаемых нами детей 6 были ранее оперированы, причем 2 пациента — дважды: 4 — по поводу острого аппендицита, 2 — по поводу кишечной непроходимости, 1 — по поводу перитонита, 1 — острого холецистита. Как правило, в медицинской документации таких больных отмечается наличие «катарального аппендицита» и необходимость хирургического вмешательства не вызывает сомнений. Довольно типично то, что, со слов родителей, врачи, оперировавшие ребенка, в частных беседах отрицали действительное наличие аппендицита или перитонита.
Длительность лихорадочного и абдоминального вариантов ПБ обычно составляет от 1 до 3 дней, реже удлиняется до 1–2 нед.
Перитонит, как и суставной синдром, наиболее закономерен для детского возраста.
Суставной синдром характеризуется артралгиями, воспалением крупных суставов. Артриты и артралгии по различным данным наблюдаются в 35–80% случаев, причем в 17–30% они являются первыми признаками заболевания. В момент приступа появляются внезапные суставные боли в одном или нескольких суставах, которые могут сопровождаться отеком, гиперемией и гипертермией суставов. Продолжительность суставного варианта приступа ПБ составляет 4–7 дней, иногда удлиняясь до 1 мес. В отличие от изолированной лихорадки или пароксизмального перитонита при этом варианте ПБ артралгии часто сохраняются и после приступа, постепенно стихая в течение нескольких месяцев. Неспецифичность клинической картины при суставном варианте ПБ приводит к тому, что у больных диагностируют ревматоидный артрит, ревматизм, системную красную волчанку и пр. Отец одной из наших пациенток, по национальности армянин, длительные годы наблюдался с диагнозом «ревматоидный артрит», и только при выявлении ПБ у ребенка нами был генетически установлен тот же диагноз у него.
Торакальный вариант с плевральным синдромом встречается реже — около 40% случаев, изолированно — в 8%, в сочетании с абдоминальным синдромом — в 30%. При торакальном варианте развивается одно-двусторонний плеврит со стерильным выпотом. Длительность этого синдрома — 3–7 дней. Как правило, таким больным ошибочно устанавливается диагноз плеврита или плевропневмонии.
Кожные изменения во время приступа ПБ встречаются в 20–30% случаев. Наиболее типичной является рожеподобная сыпь, но могут встречаться пурпурные высыпания, везикулы, узелки, ангионевротические отеки. Иногда клинически ПБ протекает подобно аллергической реакции вплоть до отека Квинке и крапивницы.
Другими проявлениями ПБ могут быть головная боль, асептический менингит, перикардит, миалгия, гепатолиенальный синдром, острый орхит.
Среди наших пациентов у 12 ПБ протекала по абдоминальному варианту, у 3 — по абдоминально-суставному. 11 из них поступали в РДКБ с другими диагнозами: хронический холецистит, панкреатит, гастродуоденит, болезнь Крона, колит неясной этиологии, ревматоидный артрит, СКВ (системная красная волчанка), хронический гломерулонефрит и только 4 — с направляющим диагнозом «периодическая болезнь». Большинство больных поступают в отделение гастроэнтерологии с жалобами на рецидивирующие абдоминальные боли, при вовлечении почек с развитием протеинурии и нефротического синдрома — в отделение нефрологии, при немотивированной рецидивирующей лихорадке — в инфекционные и диагностические отделения.
Манифестация заболевания может приходиться на различный возраст. Описаны случаи довольно позднего манифестирования ПБ, после 20–25 лет. По нашим наблюдениям, у большинства больных первый приступ ПБ отмечался в возрасте 2–3 лет (9 пациентов), у 1 — с рождения, у 2 — в 0,5–1,5 года, у 2 — в 4–5 лет, у 1 — в 11–12 лет.
Частота и периодичность приступов варьируются у разных больных в широких пределах: от нескольких раз в неделю до 1–2 раз в несколько лет. У большинства больных приступы имеют довольно стабильный ритм. Однако в литературе описаны случаи, когда приступы могли прекратиться на несколько лет или, наоборот, возобновиться после длительного перерыва под воздействием внешних факторов (смена места жительства, женитьба или замужество, рождение ребенка, служба в армии и др.). У наших пациентов периодичность приступов была довольно постоянной: у 1-го — 2 раза в нед, у 4-х — 1 раз в нед, у 5-ти — 1 раз в 2–3 нед, у 2-х — 1 раз в мес, у 1 — 1 раз в 2–3 мес, у 1-го — 1 раз в 6–12 мес.
Через некоторое время от начала манифестации у большинства больных отмечается гепатомегалия, которая, по нашим наблюдениям, может варьироваться от +1 до +5 см. Постепенно развивается и спленомегалия, величина которой у некоторых пациентов достигала +7 см. Однако увеличение печени и селезенки выявляется не у всех больных. Очевидно, эти процессы зависят от частоты и количества перенесенных приступов и развития амилоидоза.
Амилоидоз как осложнение периодической болезни
Каждый приступ ПБ сопровождается выбросом большого количества медиаторов, образованием воспалительных белков. Из тканей и серозных покровов эти белки попадают в кровь, где длительно циркулируют. Таким образом, перед организмом стоит задача каким-то образом элиминировать эти белковые вещества. Чем чаще и выраженнее приступы ПБ, тем острее проблема утилизации. Одним из способов избавиться от избытка циркулирующих белковых молекул является их переработка с образованием нерастворимого белка — амилоида. Выражаясь образно, амилоид — это плотно упакованный белковый «мусор». Образование и отложение в тканях амилоида приводит к развитию амилоидоза.
Амилоидоз (от лат. amylum — крахмал) — собирательное понятие, включающее группу заболеваний, характеризующихся внеклеточным отложением белков в виде характерных амилоидных фибрилл. Эти нерастворимые фибриллярные белки могут быть локализованы в одном специфическом месте или могут быть распространены в различных органах, в том числе таких жизненно важных, как почки, печень, сердце и др. Такое накопление приводит к органной дисфункции, недостаточности органа и в конечном итоге смерти.
Структура амилоида идентична при всех его типах и представляет собой жесткие неразветвляющиеся фибриллы диаметром около 10 нм, обладающие складчатой β-кросс-конформацией, благодаря которой возникает эффект двойного лучепреломления в поляризованном свете при окраске Конго-красным. Окраска щелочным Конго-красным является наиболее распространенным и доступным методом выявления амилоида.
Амилоид состоит из фибриллярных белков (фибриллярный компонент, F-компонент) и гликопротеидов плазмы крови (плазменный компонент, P-компонент). Предшественники F-компонента различаются при различных видах амилоидоза (на сегодняшний день известно до 30 белков-предшественников, они определяют тип амилоидоза); предшественник Р-компонента один — сывороточный амилоидный Р-компонент (SAP), схожий с α-глобулином и СРБ.
Фибриллы амилоида и плазменные гликопротеиды образуют комплексные соединения с хондроитинсульфатами ткани с участием гематогенных добавок, среди которых основными являются фибрин и иммунные комплексы. Связи между белковыми и полисахаридными составляющими в амилоидном веществе особо прочные, что объясняет отсутствие эффекта при воздействии на амилоид различных ферментов организма, т. е. амилоид нерастворим.
При ПБ основой формирования фибриллярного компонента амилоида является сывороточный острофазовый белок SAA. SAA является a-глобулином, близким по своим функциональным свойствам к СРБ. SAA синтезируется клетками разных типов (нейтрофилами, фибробластами, гепатоцитами), его количество повышается во много раз при воспалительных процессах и опухолях. У человека выделено несколько типов SAA, и только фрагменты некоторых из них входят в состав амилоидных фибрилл, что, возможно, объясняет развитие амилоидоза только у части больных, несмотря на повышенную выработку SAA. Из сывороточного SAA-предшественника в тканях образуется АА-белок (белок амилоида А), который и является основой амилоидных фибрилл. Поэтому тип амилоидоза, развивающегося при ПБ, называется АА-амилоидоз.
Таким образом, основой развития амилоидоза при ПБ является избыточное образование белка-предшественника SAA. Но для образования амилоидного белка необходимы клетки, которые будут его синтезировать — амилоидобласты. Эту функцию выполняют в основном макрофаги-моноциты, а также плазматические клетки, фибробласты, ретикулоциты и эндотелиальные клетки. Макрофаги перерабатывают АА-белок в полноценные амилоидные фибриллы на своей поверхности и откладывают его в межуточной ткани. Поэтому наибольшее накопление амилоида при ПБ отмечается в органах, где макрофаги занимают фиксированное положение: почки, печень, селезенка. Постепенно все увеличивающиеся отложения амилоида приводят к сдавливанию и атрофии паренхиматозных клеток, склерозу и недостаточности органа.
Амилоидоз при ПБ болезни развивается по различным данным у 10–40% больных. Некоторые пациенты, несмотря на довольно частые приступы, не развивают амилоидоз вовсе. Вероятно, развитие амилоидоза зависит от особенностей строения белка-предшественника у данного пациента и генетической способности макрофагов синтезировать амилоид.
Несмотря на то, что амилоидоз может развиваться в любом органе и ткани, амилоидное поражение почек играет определяющую роль для прогноза и жизни больного ПБ. При развитии АА-амилоидоза почки поражаются в 100% случаев.
В почках роль амилоидобластов выполняют мезангиальные и эндотелиальные клетки.
В процессе отложения амилоида в почечной ткани и вызванного им поражения органа можно проследить определенную стадийность. Выделяют 4 стадии амилоидоза почек: латентную (диспротеинемическую), протеинурическую, нефротическую (отечную) и уремическую (азотемическую).
В латентную стадию изменения в почках незначительны. Отмечаются нарушения гломерулярного фильтра в виде очагового утолщения, двухконтурности мембраны и аневризм ряда капилляров. В гломерулах амилоида нет или он обнаруживается не более, чем в 25% клубочков.
Ведущим в патогенезе этой стадии амилоидоза является значительный синтез и повышение в плазме крови концентрации белков-предшественников амилоидоза, т. е. диспротеинемия. Клинически у детей на фоне приступов ПБ может развиваться гипохромная железодефицитная анемия, гиперпротеинемия, диспротеинемия с увеличением глобулинов α2, β и γ, отмечается высокое содержание фибриногена и сиалопротеинов. Характерны увеличение и уплотнение печени и селезенки.
Изменения в моче поначалу отсутствуют или носят транзиторный характер, однако со временем протеинурия становится постоянной и более выраженной, часто наблюдается микрогематурия и цилиндрурия. Появление постоянной протеинурии характеризует переход во вторую, протеинурическую, стадию.
В протеинурической стадии амилоид появляется не только в пирамидах, но и в половине клубочков почек в виде небольших отложений в мезангии, отдельных капиллярных петлях, а также артериолах. Отмечается выраженный склероз и амилоидоз стромы, сосудов, пирамид и интермедиарной зоны, что приводит к атрофии многих глубокорасположенных нефронов.
Продолжительность этой стадии, как и предыдущей, колеблется от нескольких месяцев до многих лет. По мере нарастания тяжести амилоидоза усугубляются лабораторные показатели выраженной активности процесса: значительная протеинурия и диспротеинемия, гиперфибриногенемия, СРБ, гиперкоагуляция. Дальнейшее отложение амилоида в почечной ткани и нарастающая протеинурия приводят к развитию отечного синдрома, появление которого свидетельствует о переходе заболевания в третью, отечную, стадию.
В отечную (нефротическую) стадию амилоидоза количество амилоида в почках увеличивается. Пораженными оказываются более чем 75% гломерул. Прогрессирует склероз интерстиция и сосудов, в пирамидах и интрамедиарной зоне склероз и амилоидоз имеют выраженный диффузный характер.
Клинически эта стадия амилоидоза представлена полным нефротическим синдромом, хотя иногда может наблюдаться неполный (безотечный) нефротический синдром. Протеинурия становится массивной и, как правило, неселективной; нарастают циллиндры. Гематурия бывает редко и, как правило, незначительна. Нарастают гепатоспленомегалия, гипопротеинемия, усиливаются диспротеинемия с дальнейшим повышением уровня α1-, α2-, и γ-глобулинов, гиперфибриногенемия, гиперлипемия. Со временем появляется артериальная гипертензия, нарастает азотемия, прогрессирует почечная недостаточность.
Уремическая (азотемическая) стадия развивается в финале заболевания. В связи с нарастающим амилоидозом и склерозом наблюдаются гибель большинства нефронов, их замещение соединительной тканью, развивается ХПН (хроническая почечная недостаточность).
Клиническими особенностями ХПН при амилоидозе, отличающими ее от ХПН вследствие других заболеваний, является сохранение нефротического синдрома с массивной протеинурией, часто определяются большие размеры почек, характерно развитие гипотензии.
Часто выражен ДВС-синдром (синдром диссеминированного внутрисосудистого свертывания крови) в виде пурпуры, носовых, желудочных и кишечных кровотечений. Возможны тромбозы почечных сосудов с развитием инфарктов ишемического или геморрагического типа.
Мы наблюдали развитие амилоидоза у 4-х детей с ПБ (26% наблюдаемых больных). Транзиторная протеинурия у них появлялась на 7–8 год от манифестации заболевания, через 2–3 года она принимала постоянный характер. У 2-х детей через 1,5–2 года после установления постоянной протеинурии развился нефротический синдром, который у одного ребенка перерос в ХПН.
С момента развития нефротического синдрома детям диагностировали хронический гломерулонефрит и назначали соответствующее лечение глюкокортикоидами, которое не имело эффекта. В дальнейшем заболевание расценивали как СКВ и гормонорезистентый вариант гломерулонефрита, дети получали терапию цитостатиками, также без эффекта. Диагноз «периодическая болезнь, амилоидоз почек» в обоих случаях был впервые установлен в РДКБ.
Развитие амилоидоза в определенной степени зависит от количества перенесенных ребенком приступов ПБ. Среди наших пациентов амилоидоз почек выявлялся у тех, кто перенес более 130–150 приступов, тогда как у детей с меньшим количеством приступов признаков амилоидоза и поражения почек не отмечалось. Причем дети с нефротическим синдромом перенесли наибольшее число приступов — около 240 и 260. Следует отметить, что подобная закономерность не является абсолютной и амилоидоз может развиться и при меньшем количестве приступов ПБ.
Диагностика периодической болезни и амилоидоза
При типичном течении периодической болезни ее диагностика не представляет трудностей. Наибольшая проблема заключается в незнании большинством врачей этой патологии, что приводит к плохой выявляемости даже при наличии симптомов.
Диагностика ПБ основывается на 5 пунктах.
Диагностика АА-амилоидоза представляет значительную трудность. В большинстве случаев АА-амилоидоз своевременно не диагностируется, даже когда имеются клинические признаки заболевания. Причиной этого являются, с одной стороны, неспецифичность симптомов заболевания, а с другой — отсутствие у большинства врачей настороженности в отношении амилоидоза, что связано в том числе с его малой распространенностью у детей. Однако наши представления о частоте амилоидоза у детей ошибочны, и выявляемые случаи представляют собой лишь «вершину айсберга». Как показывают последние исследования, проведенные у взрослых пациентов, амилоидоз при жизни не диагностирован у 83% больных.
При постановке диагноза ПБ в большинстве случаев у врача возникает настороженность в отношении амилоидоза. Но часто первые подозрения на АА-амилоидоз могут возникнуть у педиатра при лечении больных с нефротическим синдромом, резистентных к стандартной глюкокортикоидной терапии.
Только изучение материалов биопсии с обязательным окрашиванием Конго-красным и поляризационной микроскопией позволяет поставить окончательный диагноз АА-амилоидоза. Помимо этого для диагностики можно использовать специфические антитела к АА-фибриллам. Наиболее достоверной является биопсия почки. Частота выявления АА-амилоидоза в этом случае достигает 90–100%. Чем более распространен процесс, тем больше вероятность выявления АА-амилоида в других местах (желудочно-кишечный тракт (ЖКТ) — слизистая и подслизистая, слизистая десны, прямая кишка, жировая биопсия). Наиболее информативной среди непочечных биопсий является биопсия стенки ЖКТ и прямой кишки, при которой вероятность выявления амилоида составляет 50–70%.
Лечение
При периодической болезни основой терапии является назначение колхицина. Колхицин обладает антимитотическим эффектом в отношении амилоидобластов при периодической болезни — макрофагов и стабилизирует мембрану нейтрофилов, препятствуя выбросу пирина. Колхицин назначается пожизненно в дозе 1–2 мг/сут. Он хорошо переносится, иногда возникают диспептические явления, которые не требуют полной отмены препарата. Колхицин в большинстве случаев полностью предотвращает появление приступов ПБ или значительно снижает их частоту и выраженность, предотвращает развитие амилоидоза почек, снижает выраженность его проявлений. При почечной недостаточности дозу снижают исходя из степени снижения клубочковой фильтрации. Препарат может быть временно отменен при острых инфекциях у ребенка.
Нами наблюдался мальчик, который был направлен в РДКБ с генетически установленным диагнозом периодической болезни в возрасте 16 лет. Приступы ПБ отмечались у него с 4-летнего возраста, протекали с периодичностью 1 раз в 2–3 нед в виде лихорадки с абдоминальным болевым синдромом, 1–2-кратной рвотой, головной болью и выраженной слабостью. Приступы длились около суток, затем в течение 1–2 дней была столь выраженная слабость, что мальчик не мог встать с постели, не посещал школу. Признаков амилоидоза не выявлялось.
В РДКБ ребенку был назначен колхицин в дозе 2 мг/сут. За последующие 2 года наблюдения число приступов резко снизилось до 1–2 раз в год, а в течение последних 10 месяцев не было ни одного. Сейчас юноша успешно обучается в вузе, живет в общежитии в другом городе, чувствует себя хорошо.
В лечении ПБ и профилактике амилоидоза необходима организация правильного питания ребенка. Увеличение общего количества белка в рационе стимулирует амилоидогенез, тогда как белок печени и мышцы сердца ингибируют его. Рекомендуется диета со сниженным на 50% содержанием животного (особенно казеина) и растительного белков и увеличением продуктов, содержащих крахмал. Диета должна быть достаточно обогащенной фруктами, овощами и другими шлакогонными продуктами. Белок предпочтительнее давать ежедневно (100 г печени, сырой или кулинарно обработанной). Печень употребляют годами, в виде повторных многомесячных курсов. Используют гепатотропные препараты повторными курсами: по 2–4 мес Эссенциале, Липоевую кислоту.
Прогноз
При своевременной постановке диагноза и назначении колхицина прогноз ПБ благоприятен.
При отсутствии терапии наибольшую опасность представляет развитие почечного амилоидоза, который, по сути, является единственной причиной смерти больных с ПБ. Анализ заболеваемости у взрослых и детей показывает, что при естественном течении периодической болезни приблизительно у 50% больных терминальная стадия почечной недостаточности развивается через 5 лет от момента появления протеинурии, у 75% — в течение 10 лет.
По вопросам литературы обращайтесь в редакцию.
А. В. Малкоч, кандидат медицинских наук
РГМУ, Москва



