Что такое равновесная концентрация фармакология
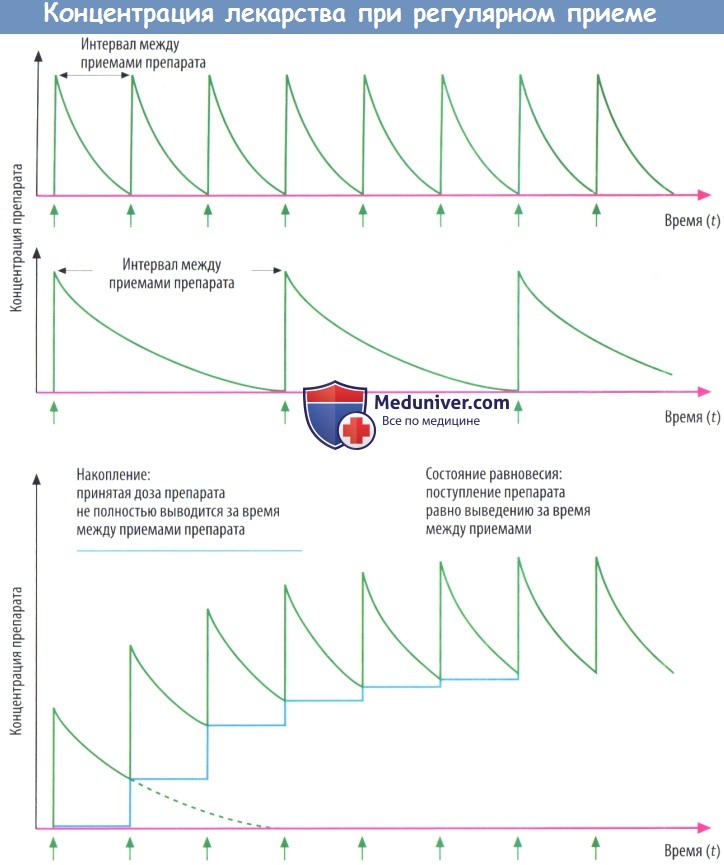
а) Концентрация препарата в крови при повторном введении. Если больной принимает препарат в течение длительного периода через одинаковые промежутки времени, подъем и падение концентрации лекарственного вещества в крови определяется взаимоотношением между t1/2 и временным интервалом между дозами.
В том случае, если количество препарата, поступившего в организм, элиминируется прежде, чем принята следующая доза, концентрация вещества при повторных приемах через равные промежутки времени будет постоянной.
Если препарат поступает в организм прежде, чем предыдущая доза полностью элиминируется, следующая доза «добавляется» к остаточному количеству препарата, все еще присутствующему в организме, т. е. вещество накапливается.
Чем короче интервал между дозами по отношению к t1/2, тем выше остаточное количество препарата, к которому прибавляется очередная доза, и тем активнее препарат накапливается в организме.
Однако при фиксированном интервале межу приемами препарат не накапливается бесконечно, в итоге наступает состояние равновесия (концентрация Css), или равновесное накопление. Так происходит потому, что активность процессов элиминации зависит от концентрации. Чем выше концентрация препарата, тем большее его количество элиминируется за единицу времени.
После приема нескольких доз концентрация достигает уровня, при котором количество препарата, выведенного и поступившего в организм за единицувремени, становится одинаковым, т. е. достигается состояние равновесия.
В пределах этого диапазона концентрации уровень вещества в крови продолжает повышаться (пик) и падать (низшая точка) в процессе приема лекарственного средства через равные промежутки времени.
Максимальное значение равновесной концентрации (Css) зависит от количества препарата (D), принятого за время между введениями (τ), и клиренса (Cl): C55 = D/(τ x Cl).
Скорость, с которой достигается равновесное состояние, соответствует скорости элиминации препарата. Время, необходимое для достижения 90%-го плато концентрации, примерно в 3 раза превышает t1/2 элиминации.
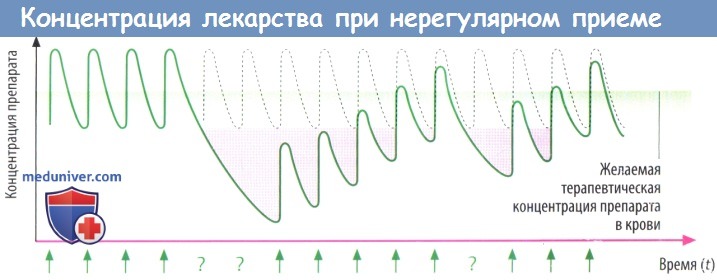
б) Концентрация препарата в крови при приеме через неравные промежутки времени. На практике бывает трудно добиться такого уровня препарата в крови, который колебался бы равномерно около желаемой эффективной концентрации. Например, если пропущены две дозы подряд, концентрация препарата в крови падает ниже терапевтической, и требуется больше времени, чтобы она вновь повысилась до желаемого уровня.
Нередко пациенты не соблюдают режим приема препарата (приверженность к лечению — строгое выполнение рекомендаций врача).
В результате концентрация препарата в крови в предутренние часы может опускаться ниже желаемой, а порой и крайне необходимой.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
ОСНОВНЫЕ ВОПРОСЫ ФАРМАКОКИНЕТИКИ




![]()
![]()
Фармакокинетика — раздел клинической фармакологии, предметом которого является изучение процессов всасывания, распределения, связывания с белками, биотрансформации и выведения лекарственных веществ. Ее развитие стало возможным благодаря разработке и внедрению в практику высокочувствительных методов определения содержания лекарственных веществ в биологических средах — газожидкостной хроматографии, радиоиммунных, ферментно-химических и других методов, а также благодаря разработке методов математического моделирования фармакокинетических процессов. На основании данных о фармакокинетике того или иного препарата определяют дозы, оптимальный путь введения, режим применения препарата и продолжительность лечения. Регулярный контроль содержания лекарственных средств в биологических жидкостях позволяет своевременно корригировать лечение.
Фармакокинетические исследования необходимы при разработке новых препаратов, их лекарственных форм, а также при экспериментальных и клинических испытаниях ЛС.
Процессы, происходящие с лекарственными препаратами в организме, могут быть описаны с помощью ряда параметров.
Одним из основных показателей, определяющих фармакологический эффект, считают концентрацию ЛС на уровне рецептора, однако в условиях целостного организма установить её невозможно. В эксперименте было доказано, что в большинстве случаев между концентрацией препарата в крови и его содержанием в области рецептора существует корреляция.
В связи с этим для определения фармакокинетических параметров изучают содержание ЛС в крови. Для того чтобы получить соответствующее представление о поступлении препарата в кровь и выведении его из организма, отслеживают изменения концентрации ЛС в плазме крови на протяжении длительного времени. Содержание препаратов в плазме крови определяют методами жидкостной или газожидкостной хроматографии, с помощью радиоиммунного или иммуноферментного анализа и другими способами.
Такой график носит название фармакокинетической кривой (рис. 1).
 Время после введения
Время после введения
Константы скорости элиминации (Кel), абсорбции (Ка) и экскреции (Кex) – характеризуют соответственно скорость исчезновения препарата из организма путем биотрансформации и выведения, скорость поступления его из места введения в кровь и скорость выведения с мочой, калом, слюной и др.
Период полувыведения (Т1/2) — время, необходимое для уменьшения вдвое концентрации препарата в крови, зависит от константы скорости элиминации (Т1/2= 0,693/Кel).
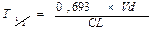
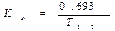
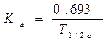
Распределение препарата в организме характеризуют период полураспределения, кажущаяся начальная и стационарная (равновесная) концентрации, объем распределения.
Период полураспределения (Т1/2,a) — время, необходимое для достижения концентрации препарата в крови, равной 50% от равновесной, т.е. при наличии равновесия между кровью и тканями.
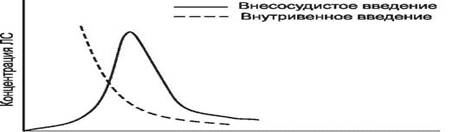
Кажущаяся начальная концентрация (С0) — концентрация препарата, которая была бы достигнута в плазме крови при внутривенном его введении и мгновенном распределении по органам и тканям.
Равновесная концентрация (Сss) — концентрация препарата, которая установится в плазме (сыворотке) крови при поступлении препарата в организм с постоянной скоростью. При прерывистом введении (приеме) препарата через одинаковые промежутки времени в одинаковых дозах выделяют максимальную (Сssmax) и минимальную (Сssmin) равновесные концентрации.

Общий клиренс препарата (Clt) характеризует скорость “очищения” организма от лекарственного препарата.
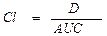 где Сl – общий клиренс; D – доза введенного препарата; AUC – площадь под фармакокинетической кривой.Выделяют почечный (Clr) и внепочечный (Cler) клиренсы, которые отражают выведение лекарственного вещества соответственно с мочой и другими путями (прежде всего с желчью). Общий клиренс является суммой почечного и внепочечного клиренса.
где Сl – общий клиренс; D – доза введенного препарата; AUC – площадь под фармакокинетической кривой.Выделяют почечный (Clr) и внепочечный (Cler) клиренсы, которые отражают выведение лекарственного вещества соответственно с мочой и другими путями (прежде всего с желчью). Общий клиренс является суммой почечного и внепочечного клиренса. 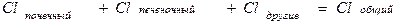
Абсолютная биодоступность (f) — часть дозы препарата (в %), которая достигла системного кровотока после внесосудистого введения, равна отношению AUC после введения исследуемым методом (внутрь, в мышцу и др.) к AUC после внутривенного введения. Относительную биодоступность определяют для сравнения биодоступности двух лекарственных форм для внесосудистого введения. Она равна отношению (AUC’/AUC)(D/D’) после введения двух сравниваемых форм. Общая биодоступность — часть принятой внутрь дозы препарата, которая достигла системного кровотока в неизмененном виде и в виде метаболитов, образовавшихся в процессе всасывания в результате так называемого пресистемного метаболизма, или “эффекта первичного прохождения”.
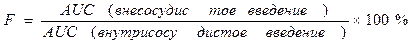
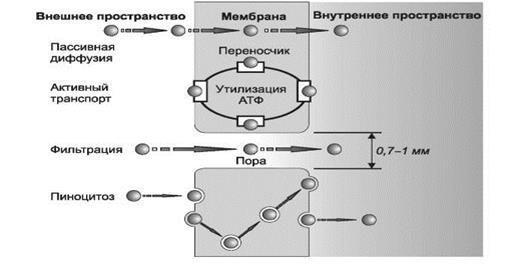
ВСАСЫВАНИЕ — процесс поступления лекарственного вещества из места введения в кровь. Существуют четыре механизма всасывания ЛС при энтеральном введении (рис. 2):
Ø пассивная диффузия;
Ø активный транспорт;
Ø фильтрация через поры;
Прохождение большинства лекарственных препаратов через слизистую оболочку пищеварительного тракта определяется их растворимостью в липидах и ионизацией. При приеме лекарственных веществ внутрь скорость их абсорбции отличается в различных отделах ЖКТ.
После прохождения через стенку желудка и/или кишечника лекарственный препарат поступает в печень. Некоторые лекарственные вещества под влиянием ферментов печени подвергаются значительным изменениям (“эффект первичного прохождения”). Именно поэтому, а не вследствие плохой абсорбции, для достижения достаточного эффекта дозы некоторых препаратов (пропранолола, аминазина, опиатов) при приеме их внутрь должны быть значительно больше, чем при внутривенном введении. Биотрансформацию вещества при первичном прохождении через печень в процессе всасывания называют пресистемным метаболизмом. Интенсивность пресистемного метаболизма зависит от скорости тока крови в печени.
На процесс всасывания лекарств в желудке и кишечнике оказывает влияние рН, который в желудке равен 1-3, в двенадцатиперстной кишке — 5-6, а в тонкой и толстой кишках — около 8. Кислоты легче всасываются в желудке, а основания — в тонкой или толстой кишке.
Под действием кислой среды желудка некоторые лекарственные средства, в частности бензилпенициллин, могут разрушаться.
На лекарственные препараты оказывают также действие ферменты желудочно-кишечного тракта, которые способны инактивировать белки и полипептиды (АКТГ, вазопрессин, инсулин и т.д.), а также некоторые другие вещества (прогестерон, тестостерон, альдостерон). Соли желчных кислот в свою очередь могут ускорить всасывание лекарственных средств или замедлить его при образовании нерастворимых соединений.
На всасывание лекарственных веществ влияют также моторика желудочно-кишечного тракта, объем и состав пищи, количество принимаемой жидкости, интервал времени между едой и приемом препаратов. Так, молоко нарушает всасывание тетрациклинов, ампициллина и амоксициллина. Следует учитывать и стимулирующее действие пищи на секрецию желудочного сока и соляной кислоты.
Для переноса веществ в ЖКТ особое значение имеют большая площадь поверхности кишечника и влияние постоянного кровотока в слизистой оболочке на градиенты концентрации между просветом кишечника и кровью. Путем диффузии и осмоса через слизистую оболочку кишечника переносятся, в частности, вода, С1 ¯, а также такие вещества, как аскорбиновая кислота, пиридоксин и рибофлавин. Поскольку клеточные мембраны содержат большое количество липидов, для диффузии через мембрану вещества должны быть в некоторой степени жирорастворимыми. Согласно теории неионной диффузии, указанным путем переносятся главным образом недиссоциированные соли слабых кислот или слабых оснований. Это необходимо учитывать при назначении лекарств, большая часть которых всасывается путем диффузии. Для переноса какого-либо вещества в соответствии с уравнением Гендерсона-Гассельбаха особое значение имеет рКа этого вещества и рН в просвете кишечника:
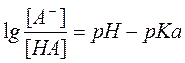 ,
, 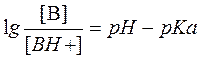 , где
, где
[А¯], [ВН + ] – молярные концентрации ионизированных,
[НА], [В] – неионизированных форм кислоты НА и основы В;
рН – кислотно-основной показатель среды;
рКа – логарифм константы диссоциации соединения, количественно равный значению рН, при котором анализируемое соединение диссоциирует наполовину.
Таким образом, факторы, влияющие на процессы всасывания ЛВ, разнообразны: растворимость вещества в липидах, степень ионизации молекулы (чем меньше ионизированная молекула, тем лучше она всасывается), перистальтика кишечника, характер и количество пищевой массы, особенности регионарного кровотока, состояние соединительной ткани, агрегантное состояние веществ, сочетание лекарственных средств.
Что такое равновесная концентрация фармакология

Результаты исследований локального постинъекционного действия растворов лекарственных средств на живые ткани человека и животных показали важную роль концентрации и других физико-химических показателей качества растворов в формировании ятрогенного воспаления и повреждения тканей в месте инъекции. [3, 5, 9]. Было установлено, что подкожные и внутримышечные инъекции высококонцентрированных лекарственных растворов, величина концентрации которых превышала 10 %, чаще приводили к образованию воспалительных инфильтратов. Также обнаружено, что развитие флебитов и тромбозов подкожных вен пациентов может быть обусловлено локальным действием высококонцентрированных растворов при их длительном и многократном внутривенном введении [4, 6]. Микроскопические исследования мазков крови после смешивания ее с растворами лекарственных средств, имеющих разные показатели концентрации активного вещества также показали наличие агрессивных свойств высококонцентрированных растворов на клетки крови [2].
Таким образом, высокая концентрация может придавать растворам лекарственных средств агрессивные раздражающие свойства по отношению к инъецируемым тканям, вызывая развитие в них воспаления и повреждения, вплоть до необратимого повреждения [1, 7]. Однако, было замечено, что раздражающим действием на ткани могут обладать растворы не только с высокими, но и с низкими показателями концентрации активного вещества, значение которой указаны на ампуле с раствором лекарственного средства, например растворы нестероидных противовоспалительных средств [8, 10]. Поскольку величина концентрации, указанная на ампуле или флаконе с раствором отражает лишь содержание активного инградиента в единице объема, то изучение концентрации вспомогательных ингредиентов в готовом растворе и определение их роли в формировании суммарной концентрации и осмотической активности раствора является актуальным.
Цель исследования – изучить влияние концентрации активных и вспомогательных инградиентов готовых растворов лекарственных средств на их активность.
Материалы и методы исследования
Проведено изучение состава и свойств растворов нестероидных противовоспалительных лекарственных средств, предназначенных для инъекций. Показатели концентрации действующих и вспомогательных веществ, входящих в состав растворов для инъекций, определяли по данным Паспортов лекарственных средств. С помощью осмометра марки VAPRO 5600 (USA) были исследованы показатели осмотической активности лекарственных растворов. В качестве контроля был использован раствор 0,9 % натрия хлорида. Для оценки биологической активности лекарств в экспериментах на 10 здоровых 2-месячных поросятах породы ландрас изучена динамика изменений температуры и спектра инфракрасного излучения кожи над в области медикаментозных инфильтратов, образованных подкожным введением в области передней брюшной стенки растворов нестероидных противовоспалительных лекарственных средств разной концентрации в объеме 0,5 мл. В качестве контроля были использованы значения температуры кожи над поверхностью инфильтрата, образованного подкожным введением 0,5 мл раствора 0,9 % натрия хлорида. Растворы лекарственных средств имели показатели температуры + 24 ± 0,8 ºС. Состояние поверхности кожи поросят в области инъекции оценивали в видимом и инфракрасном спектре излучения с помощью тепловизора марки ThermoTracer TH9100XX (NEC, USA) в диапазоне температур от + 25 до + 36 °С с последующей обработкой полученной информации с применением программ Thermography Explorer и Image Processor.
С помощью статистической программы BIOSTAT на персональном компьютере Lenovo R60 (USA) вычисляли среднюю арифметическую (M), ошибку средней арифметической (m), коэффициент достоверности (±). Степень различий показателей определяли в каждой серии по отношению к исходным показателям в контрольной серии. Разницу значений считали достоверной при Р ≤ 0,05.
Результаты исследования и их обсуждение
Для исследования нами были выбраны растворы нестероидных противовоспалительных лекарственных средств с разными показателями концентрации активного вещества. Ими оказались растворы нестероидных противовоспалительных средств, а именно 5 % раствор для инъекций Кетопрофен® (ОАО «Синтез», г. Курган, Россия), 3 % раствор для инъекций Кеторол® (Д-р Редди̕с Лабораторис Лтд., г.Хайдерабад, Андхра Прадеш, Индия) и 50 % раствор для инъекций Анальгин (ОАО «Ереванская химико-фармацевтическая фирма», г. Ереван, Республика Армения).
Результаты наблюдений за изменением температуры и спектра ифракрасного излучения поверхности кожи поросят в области инъекций показали, что инфильтрат, образованный подкожным введением 0,9 % натрия хлорида не вызывал развития локальной гипертермии на протяжении 60 минут после инъекции, а восстановление исходной температуры после первоначального охлаждения, вызванного введением «холодного» раствора, наступало через 5-7 минут. В то же время, растворы выбранных для исследования лекарственных средств вызывали локальное повышение температуры кожи в области инъекции на 0,3-1,8 °С, при этом локальная гипертермия регистрировалась на экране тепловизора в течении 15-60 минут после инъекции. Таким образом, все исследуемые растворы лекарственных средств, независимо от величины концентрации активного вещества, проявили раздражающие действие для мягкие ткани передней брюшной стенки, вызвав в них развитие воспаления, проявляющееся в первую очередь локальной гипертермией.
Исследования осмотической активности указанных растворов показали, что 50 % раствор анальгина имею величину осмолярности 4638 ± 12,5*мОсм/л воды, 5 % кетопрофен – 4767 ± 11,5*мОсм/л воды, а 3 % кеторол – 2971 ± 9,8*мОсм/л воды (достоверно при Р ≤ 0,05, n = 5 по сравнению с контролем), свидетельствующие о том, что низкая концентрация действующего вещества, указанная на ампуле или флаконе с раствором лекарственного средства, не исключает наличия у раствора высокой суммарной осмотической активности.
Дальнейший анализ паспортов и инструкций к применению данных лекарственных средств показал, что в состав изучаемых растворов входит от 2 до 7 различных вспомогательных веществ (таблица).
Качественный и количественный состав растворов



