Что такое синдром апера
Синдром Апера (Apert), или акроцефалосиндактилия, является редко встречающейся аномалией, которая характеризуется мальформациями черепа, лица и конечностей и в 50% случаев сопровождается умственной отсталостью различной степени тяжести. Впервые этот синдром был описан Уитоном (Wheaton) в 1894 году и обобщен Апером (Apert) в 1906 году, который представил 9 клинических наблюдений этой патологии.
По некоторым данным частота встречаемости синдрома Апера (Apert) составляет от 0,0625 до 0,1 на 10 000 родов. Вследствие высокого уровня неонатальной смертности ожидаемая распостраненность в общей популяции приблизительно оценивается на уровне от 5 х 10 6 до 1 х 10 5 на 10 000 новорожденных. Соотношение по половому признаку составляет 1:1.
Синдром Апера (Apert) представляет собой аутосомное нарушение с доминантным типом наследования. Большинство случаев возникает спорадически и обусловлено появлением новых мутаций. Описана связь возникновения патологии с увеличением возраста родителей.
При развитии заболевания вследствие новой мутации риск рецидива маловероятен. Если один из родителей является носителем заболевания, риск рецидива составляет 50%.
Наиболее типичными признаками синдрома Апера (Apert) являются краниосиностоз (с вовлечением коронарного шва при данном заболевании), двухсторонняя симметричная синдактилия конечностей (варежкообразные кисти и стопы) и гипоплазия срединных отделов лица. Дополнительные признаки, появляющиеся с переменной частотой, могут быть представлены:
• скелетными аномалиями, такими как уменьшенная по высоте и широкая голова; высокий «башенный» мозговой череп (акроцефалия); выступающий лоб; гипертелоризм и проптоз глазных яблок; глубокая переносица с носом, напоминающим клюв попугая; гипоплазия верхней челюсти; прогнатия;
• пороками сердца, такими как стеноз ствола легочной артерии, «верхом сидящая» аорта и дефекты межжелудочковых перегородок;
• аномалиями центральной нервной системы, такими как гидроцефалия, мальформации мозолистого тела и лимбических структур, аномалии извилин, гипоплазия белого вещества и гетеротопия серого вещества.
Имеются сообщения о пренатальной диагностике с помощью ультразвукового исследования и фетоскопии во всех триместрах беременности. Эхографическим маркером данной патологии в первом триместре также может быть увеличение толщины воротникового пространства.
Наиболее распространенными мутациями, которые вызывают синдром Апера (Apert), являются замены нуклеотидов серина на триптофан S252W и пролина на аргинин P253R, происходящие в гене рецептора 2 фактора роста фибробласта (FGFR2). При подозрении на синдром Апера (Apert) рекомендуется проведение молекулярно-генетических исследований у плода (путем биопсии ворсин хориона или амниоцентеза), а также обследование родителей, особенно в тех семьях, где заболевание регистрируется впервые.
Недавно выполненное исследование выявило, что при мутации P253R отмечаются меньшая выраженность черепно-лицевых аномалий и более значимымые деформации конечностей. Кроме того, с помощью генетических методов была выявлена связь между нарушением экспрессии генов рецепторов фактора роста кератиноцитов (KGFR) и возникновением деформаций конечностей при синдроме Апера (Apert).
В дифференциальный диагноз могут быть включены генетические синдромы, характеризующиеся наличием краниосиностоза, такие как синдромы Крузона (Crouzon), Пфайффера (Pfeiffer), Карпентера (Carpenter) и Сэтре-Чотцена (Saethre-Chotzen). В настоящее время для исключения этих аномалий могут использоваться молекулярно-генетические методы исследования.
До наступления периода жизнеспособности плода может быть предложено прерывание беременности. В более поздние сроки гестации стандартная акушерская такика не изменяется. Рекомендуется родоразрешение в специализированных перинатальных медицинских центрах.
 Синдром Апера I
Синдром Апера I
Синдром Апера
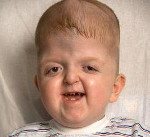
Синдром Апера – генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей. Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований. Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
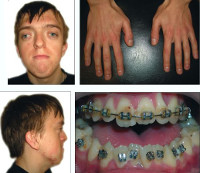
Общие сведения
Синдром Апера (акроцефалосиндактилия 1 типа) – генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания. Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее – лишь в 1995 году. Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных. Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний – акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей. Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.
Причины синдрома Апера
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной. Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями. Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R. Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния. Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности – к преждевременному зарастанию швов и остановке нормального роста черепной коробки. Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов. Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR. Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей – различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Симптомы синдрома Апера
Некоторые проявления синдрома Апера заметны с самого рождения – например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног. Среди неонатологов симптом иногда носит название «среднего пальца» – в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь. Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа. Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие. Деформации затрагивают и кости лицевого черепа – переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса. Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
У взрослых лиц, страдающих синдромом Апера, может возникать атрофия зрительных нервов вплоть до полной слепоты. Интеллектуальное развитие больных часто отстает от возрастной нормы, однако достоверно неизвестно, обусловлено это генетическими нарушениями или вторичными факторами (хронической внутричерепной гипертензией). Практически всегда при синдроме Апера наблюдается карликовый рост. При соответствующем паллиативном лечении и уходе больные могут доживать до преклонного возраста, но риск внезапной смерти из-за поражений дыхательной, нервной и сердечно-сосудистой систем у них намного выше, чем в популяции.
Диагностика
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов. При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа – башенный череп или брахикефалия, характерный внешний вид лица.
С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов – стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности. Также возможно наличие иных пороков развития – аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения.
Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию. Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния. Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера – пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение синдрома Апера
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни. Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления. По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект. Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах. Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется. При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Прогноз и профилактика
Прогноз синдрома Апера неопределенный по причине очень широкого спектра проявлений и значительного диапазона их выраженности. На прогноз также оказывают влияние такие факторы, как своевременность диагностики заболевания, объем паллиативного и симптоматического лечения. При относительно легких случаях синдрома Апера или правильной терапии этого состояния больные могут доживать до преклонного возраста. При этом возможно снижение интеллекта и появляющиеся с возрастом нарушения все новых органов и систем, что негативно сказывается на качестве жизни пациентов. В тяжелых случаях наблюдается летальный исход в раннем детстве из-за врожденных пороков сердца или полиорганной недостаточности.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа. Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком. Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Что такое синдром апера
Синонимы синдрома Апера I. Акросфеносиндактилия. Акрокраниодисфалангия. Акроцефалия. Синдром синдактилической оксицефалии.
Определение синдрома Апера I. Редкий комбинированный порок развития с акроцефалией и синдактилией.
Авторы. Apert Eugene — французский педиатр, Париж, 1868—1940. Синдром впервые описал Wheaton в 1894 г.
Симптоматология синдрома Апера I:
1. Дизостозы костей черепа: преждевременный синостоз венечного шва (акроцефалия, высокий шпилеподобиый череп), стреловидного шва (скафоцефалия, челнообразный удлиненный череп) или других швов.
2. Дисморфия лицевого черепа: гипертелоризм, широкий корень носа, щелевидный нос, плоские глазницы, экзофтальм.
3. Кожные или костные синдактилии вплоть до развития «ковшеобразной кисти», обычно двусторонние. Редко — полидактилия.
4. Ранее считались факультативными признаками луче-локтевые синостозы, синостозы больших суставов, особенно локтевого, hallux varus, пороки развития позвонков, аплазия акромиоклавикулярных суставов, высокое стояние неба, расщепленный язычок, атрезия заднепроходного отверстия, атрофия зрительного нерва, задержка психического развития, малый рост.
Этиология и патогенез синдрома Апера I. Генетически обусловленное наследственное страдание, проявляющееся на 9-й неделе эмбриональной жизни. Абортивные случаи наследуются по доминантному типу. По-видимому, известную роль могут играть и экзогенные вредные факторы, действующие в указанный эмбриональный период (ненаследственная фенокопия).
Поражение мезенхимального примордиального черепа (недостаточный рост и одновременное закрытие отдельных швов) и повышение внутричерепного давления, создающие несоответствие между патологическим черепом и нормальным ростом мозга, приводят к деформациям черепа. Часты семейные случаи заболевания. Наблюдают переходные формы с S. Crouzon и S. Greig, что, по данным Bonnevie, сближает их с так называемым кранио-дигито-фациальными аномалиями и комплексом дискранио-дисфалангий.
Дифференциальный диагноз синдрома Апера I. Синдромы черепно-нижнечелюстно-лицевой дисморфии. S. Poland.
Синдром Апера I
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
— Вернуться в содержание раздела «генетика» на нашем сайте
Синдром Апера (Apert)
Врожденное заболевание, основными проявлениями которого является деформация мозгового и лицевого черепа, сращение и недоразвитие пальцев кистей и стоп.
Также может наблюдаться недоразвитие верхней челюсти, аномалии зубов, врожденные пороки сердца, расщелина неба, аномалии желудочно-кишечного тракта и мочевыделительной сиситемы.
Предварительный диагноз ставится при рождении на основании клинической картины. Постановка окончательного диагноза устанавливается на основании детекции мутации в геноме. Известна конкретная локализация поломки — ген FGFR2 в хромосоме 10q26. Также существуют варианты пренатальной (дородовой) ДНК- диагностики.
Чем обусловлено изменение формы черепа при синдроме Apert
Синдактилия пальцев кистей и стоп, какие особенности бывают при данном заболевании?!
Выделяют три основных типа синдактилии при синдроме Apert.
При 1 типе кисть напоминает по форме лопату. При нем большой палец имеет правильную ось, или незначительное отклонение в лучевую сторону. В первом и четвертом межпальцевом промежутке как правило синдактилия неполная. Для устранения такой синдактилии пересадка дополнительной кожи чаще всего не требуется. Синдактилия между остальными пальцами является простой, что означает отсутствие костного компонента в сращении. При этом концевые отделы 2-5 пальцев могут быть не спаяны. Но не смотря на это при разделении 2-4 пальцев пересадка дополнительной кожи все равно потребуется.
При 2 типе кисть напоминает по форме ложку. В данном случае первый палец укорочен и имеет отклонение в лучевую сторону, также наблюдается тотальная (полная) синдактилия во всех межпальцевых промежутках, когда сращение доходит до концевых отделов пальцев со сросшимися ногтевыми пластинами. При этом типе сращение является простым, но часто может наблюдаться костное сращение ногтевых фаланг 3-4 пальцев. Без пересадки дополнительной кожи разделить пальцы не удастся.
При 3 типе кисть похожа на бутон. Данный тип является самым сложным в техническом плане. Первый и пятый пальцы относительно своей оси развернуты друг к другу. Сращение между всеми пальцами в области ногтевых пластин является костным, дальше пальцы расходятся веерообразно что и создает сходство с бутоном цветка.
Эти нюансы являются определяющими в тактике хирургического лечения.
Сращение пальцев стоп очень плотное, чаще костное со слиянием ногтевых пластин. При таких условиях требуется много дополнительных операций с пересадкой большого количества донорской кожи. Что в большинстве случаев не имеет смысла, поскольку синдактилии пальцев стоп редко влияют на функцию.
Основным показанием для операций на стопе является сгибательная деформация второй плюсневой кости. Это проявляется наличием выступающей « шишки » по подошвенной поверхности стопы, у основания второго пальца. Очень часто на этом месте формируется натоптыш.
Этапность лечебного процесса
Преимущественным является устранение сращения костей черепа путем установки специальных аппаратов раздвигающих кости. Своевременность оказания данного хирургического вмешательства оказывает непосредственное влияние на дальнейшее развитие центральной нервной системы и как следствие уровень интеллектуального развития.
Далее после оценки общего соматического статуса, необходимо приступать к устранению сращений пальцев кистей. В зависимости от типа синдактилий очередность хирургических вмешательств на кисти может меняться.
Но существуют общие принципы:
— Отделение и выпрямление 1 пальца играет важное значение для функции. Полноценный большой палец дает 50-70% к функции кисти. Поэтому данное вмешательство является первичным.
— Далее устранение сращения 4-5 пальцев, так как 5 палец как правило имеет более развитые межфаланговые суставы что уже даст возможность более полноценного двухстороннего схвата, чем это было бы со вторым пальцем.
— Этап операции с заимствованием донорской кожи и пересадкой на области ее потребности, является обязательным при любом типе сращения. Донорская кожа берется с области паховой складки. Швы накладываются внутрикожно, снятие швов не требуется.
Рекомендации по диспансерному наблюдению
Пациент с диагнозом синдром Apert должен наблюдаться у ряда специалистов по причине большого количества сопутствующих заболеваний:
СИНДРОМ АПЕРА: клинические проявления и этиология
Пороки развития черепно-лицевой области занимают 3-е место среди других видов врожденных аномалий. По данным экспертов Всемирной организации здравоохранения (1999), около 7% живорожденных детей имеют врожденные пороки и уродства черепно-лицевой области.
Пороки развития черепно-лицевой области занимают 3-е место среди других видов врожденных аномалий. По данным экспертов Всемирной организации здравоохранения (1999), около 7% живорожденных детей имеют врожденные пороки и уродства черепно-лицевой области. Среди врожденных черепно-лицевых деформаций около 30% приходится на краниосиностозы. Из всех синдромальных форм краниосиностозов наиболее часто встречается, по мнению подавляющего большинства специалистов, синдром Апера. В отечественной литературе, к сожалению, часто можно встретить неполную, а иногда и противоречивую информацию о данном синдроме. D. Leibek и C. Olbrich указывают следующие признаки синдрома Апера: дизостозы костей черепа, преждевременный синостоз венечного шва (акроцефалия, высокий шпилеподобный череп), стреловидного шва (скафоцефалия) или других швов; дисморфия лицевого черепа: глазной гипертелоризм, широкий корень носа, щелевидный нос, плоские глазницы, экзофтальм; кожные или костные синдактилии, обычно двусторонние; редко — полидактилия [5]. Ранее считались факультативными признаками лучелоктевые синостозы, синостозы крупных суставов, особенно локтевого, hallux varus, пороки развития позвонков, аплазия акромиоклавикулярных суставов, высокое стояние неба, расщепленный язычок, атрезия заднепроходного отверстия, атрофия зрительного нерва, задержка психического развития, малый рост.
Л. О. Бадалян в своем труде, посвященном описанию клинических проявлений различных синдромов, отмечает, что синдром Апера проявляется изменением формы головы (акроцефалия) и полисиндактилией, большие пальцы ног увеличены в размерах, имеются добавочные большие пальцы, психическое развитие не нарушено [1].
Давая клиническую характеристику синдрома Апера, Х. А. Калмакаров, Н. А. Рабухина, В. М Безруков отмечают, что у синдрома Апера, сочетающего в себе краниофациальный дизостоз с акроцефалией и синдактилией, имеется много общего с дизостозом Крузона [2]. В противоположность дизостозу Крузона, при этом виде дискраний наблюдается раннее синостозирование черепных швов. Этот процесс захватывает все черепные швы, за исключением венечных. Поэтому рост идет преимущественно в высоту, череп приобретает башенную форму и остается узким в переднезаднем и поперечном направлениях. Лоб и затылок широкие и плоские. Как и при дизостозе Крузона, отмечается выраженный экзофтальм из-за уменьшения глубины орбиты и глазной гипертелоризм из-за увеличения размеров решетчатого лабиринта. Верхняя челюсть недоразвита, соотношения зубных рядов нарушены, однако сами зубы развиваются нормально. При синдроме Апера встречается характерная деформация век — они несколько приподняты и образуют складки, поддерживающие глазные яблоки. Наблюдается также птоз верхних век и косоглазие, уплощение носа. Умственное развитие больных с этим синдромом обычно не нарушается, но отмечается очень резкая эмоциональная возбудимость. Характерно сращение нескольких пальцев верхних или нижних конечностей.
С. И. Козлова и соавторы указывают, что синдром Апера характеризуется изменениями черепа — синостоз различной выраженности в основном венечных швов в сочетании со сфеноэтмоидомаксиллярной гипоплазией основания черепа; изменениями лица — плоский лоб, глазной гипертелоризм, антимонголоидный разрез глаз; запавшая переносица, прогнатизм, полное сращение 2–5-го пальцев кистей и стоп [3].
И.Р.Лазовскис описывает синдром Апера как комплекс наследственных аномалий (аутосомно-доминантное наследование): дизостоз черепа — преждевременный синостоз венечного шва (с образованием акроцефалии), ламбдовидного шва (со скафоцефалией), часто преждевременный синостоз всех швов; дисморфия лицевого черепа: глазной гипертелоризм, расширенный корень носа, плоские орбиты, пучеглазие (экзофтальм); кожные или костные синдактилии, обычно двусторонние, реже — полидактилия; изредка наблюдаются синостоз лучевой и локтевой костей и крупных суставов, анкилоз локтевого сустава, аномалии позвоночника, высокое небо, расщепление небного язычка, офтальмоплегия, ослабление зрения; атрезия анального отверстия, умственная отсталость, карликовый рост [4].
Вся эта противоречивая информация, представленная в отечественных источниках, вносит определенную путаницу и усложняет выбор адекватного метода лечения. В основном данные, касающиеся данной темы, отражены в зарубежных источниках.
Клинические проявления синдрома Апера
Основные клинические проявления синдрома акроцефалосиндактилии, описанные французским врачом E. Apert в 1906 г. и названные его именем, сводились к следующему: краниосиностоз, гипоплазия средней зоны лица, симметричная синдактилия кистей и стоп с вовлечением 2–4-го пальцев.
В США распространенность оценивается как 1 на 65 000 (приблизительно 15,5 на 1 000 000) живорожденных. Blank описал собранный материал по 54 пациентам, рожденным в Великобритании [7]. Он разделил пациентов на две клинические категории: «типичная» акроцефалосиндактелия, к которой он применил название «синдром Апера», и другие формы, смешанные в общую группу как «нетипичные» акроцефалосиндактилии. Особенность, отличающая эти типы, — «средний палец», состоящий из нескольких пальцев (обычно 2–4-й), с единственным общим ногтем, наблюдаемый при синдроме Апера и не встречающийся в другой группе. Из этих 54 пациентов 39 имели синдром Апера. Частота синдрома Апера оценивалась им как 1 на 160 000 живорожденных. Cohen и соавторы изучили распространенность случаев рождений с синдромом Апера в Дании, Италии, Испании и частично в Соединенных Штатах [9]. Общее количество дало возможность вывести расчетную частоту рождений с синдромом Апера — приблизительно 15,5 на 1 000 0000 живорождений. Данная цифра превышает примерно вдвое результаты других исследований. Czeizel и соавторы сделали сообщение о частоте рождений больных с синдромом Апера в Венгрии, она составила 9,9 на 1 000 000 живорожденных. Tolarova и соавторы сообщили, что по результатам Калифорнийской программы мониторинга врожденных заболеваний за период с 1983 по 1993 г. было идентифицировано 33 новорожденных с синдромом Апера [26]. Данные были дополнены 22 случаями, описанными в Центре краниофациальных пороков (Сан-Франциско). Частота, определенная на основании этих данных, составила 31 случай на 12,4 млн живорожденных. Больные с синдромом Апера составляют 4,5% всех случаев краниосиностозов. Большинство случаев спорадические и являются следствием новых мутаций, однако в литературных источниках имеется описание семейных случаев с полной пенетрантностью. Weech описал мать и дочь [28], Van den Bosch, по данным Blank [7], наблюдал типичную картину у матери и сына. Rollnick описал больных отца и дочь, что явилось первым примером передачи заболевания по отцовской линии [23]. Данные факты позволяют предположить аутосомно-доминантный тип наследования.
Азиаты имеют самую высокую распространенность синдрома — 22,3 на 1 млн живорождений, испанцы, напротив, самую низкую — 7,6 на 1 млн живорождений [26]. Связь с половой принадлежностью не была выявлена ни одним из исследователей.
Синдром Апера обычно диагностируется в раннем возрасте из-за обнаружения после рождения краниосиностоза и синдактилии. Для синдрома характерно наличие первичных изменений со стороны черепа уже при рождении, однако окончательное формирование патологической формы происходит в течение первых трех лет жизни. У многих пациентов имеется затруднение носового дыхания, из-за сокращения размера носоглотки и хоан, также могут быть затруднения прохождения воздуха через трахею, из-за врожденной аномалии хрящей трахеи, что может привести к ранней смерти. Возможны головная боль и рвота — признаки увеличенного внутричерепного давления, особенно в случаях, когда в процесс вовлечено несколько швов. Генеалогический анамнез представляется не столь важным, поскольку большинство случаев рождений детей с данным синдромом являются спорадическими.
Фенотипические признаки синдрома Апера
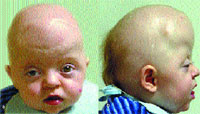 |
| Рисунок 1. Внешний вид больного в возрасте 1,5 года с синдромом Апера: анфас (а) и профиль (б) |
Черепно-лицевая область. Наиболее часто встречается коронарный краниосиностоз, приводящий к акроцефалии, брахицефалии, туррибрахицефалии. Синостозированию подвергаются также сагиттальные, ламбдовидные, лобно-основные швы. Редкая аномалия черепа в виде трилистника найдена приблизительно у 4% младенцев. Основание черепа уменьшено в размерах и часто асимметрично, передняя черепная ямка очень короткая. Передний и задний роднички увеличены в размерах и не заращены. Средняя линия свода черепа может иметь зияющий дефект, простирающийся от области глабеллы через область метопического шва до переднего родничка, через область сагиттального шва до заднего родничка. Отмечаются: глазной гипертелоризм, экзорбитизм, мелкие орбиты, нависающие надбровные дуги. Со стороны глаз наблюдаются: экзофтальм, «прерывистые брови», пальпебральные трещины, косоглазие, амблиопия, атрофия зрительного нерва, и (редко) вывих глазного яблока, снижение пигмента, врожденная глаукома, обратимая потеря зрения. Переносица часто запавшая. Нос короткий с уплощенной спинкой и с широким кончиком со стенозом или атрезией хоан, носогубные складки глубокие, возможна девиация носовой перегородки. Имеется гипоплазия средней зоны лица — верхняя челюсть гипоплазирована, скуловые дуги короткие, скуловые кости мелкие. В связи с этим отмечается относительный нижнечелюстной прогнатизм. Рот в состоянии покоя имеет трапециевидную форму. Высокое аркообразное небо, расщелина мягкого неба и язычка наблюдается в 30% случаев. Твердое небо короче, чем в норме, мягкое небо — длиннее и толще, верхнечелюстная зубная дуга имеет V-образную форму. Могут быть выступающие из ряда верхние зубы, имеющие форму совка резцы, сверхкомплектные зубы и выступающие альвеолярные гребни. Пациенты имеют низко посаженные уши и высокую вероятность снижения слуха в дальнейшем (рис. 1, 2).
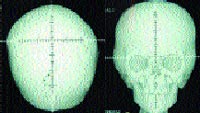 |
| Рисунок 2. Рентгенограмма черепа больного с синдромом Апера в прямой проекции (формат 3 D) |
Конечности и скелет. Одним из основных проявлений синдрома является синдактилия кистей и стоп с вовлечением 2, 3 и 4-го пальцев. Реже в процесс вовлекаются 1-й и 5-й пальцы (рис. 3). Проксимальные фаланги больших пальцев кистей и стоп укорочены, дистальные имеют трапециевидную форму. При изучении синдрома Апера Wilkie и соавторы [30] внесли изменения в классификацию синдактилий Upton (1991). При синдроме Апера центральные три пальца всегда подвергнуты синдактилии. Тип 1 — большой палец и часть 5-го пальца отделены от сросшихся пальцев; при типе 2 — только большой палец отделен от «среднего пальца»; при типе 3 — все пальцы сросшиеся. Точно так же синдактилия пальцев стопы может вовлекать три боковых пальца (тип 1), или 2–5-й пальцы с отдельным большим пальцем ноги (тип 2), или может быть непрерывной (тип 3). Cohen и Kreiborg изучили 44 пары рук и 37 пар ног пациентов с синдромом Апера, используя клинический, радиографический методы и дерматоглифику [10], а также изучили гистологические препараты верхних конечностей мертворожденного плода со сроком 31 нед. Они предположили, что различие между акроцефалосиндактилией и акроцефалополисиндактилией является ложным и что от использования этих терминов следует отказаться. Исследователи также указали на то, что при синдроме Апера патология верхних конечностей всегда более выражена, чем нижних. Сращение костей запястья с дистальными фалангами не имеет своего аналога на стопе. Возможны и другие патологические изменения конечностей: радиальное отклонение коротких и широких больших пальцев, из-за измененной проксимальной фаланги — брахидактилия; ограничение подвижности в плечевом суставе, ограниченная подвижность локтевого сустава с затруднением пронации и супинации, ограничение подвижности в коленном суставе, аплазия или анкилоз плечевого, локтевого и тазобедренного сустава. Одной из сравнительно часто встречаемых аномалий скелета при синдроме Апера является врожденное сращение позвонков. Kleiborg и соавторы обнаружили, что сращение позвонков в шейном отделе наблюдалось у 68% пациентов с синдромом Апера: единичные сращения у 37% и множественные сращения у 31% [13]. Наиболее характерно было сращение C5–C6. Напротив, сращение в шейном отделе происходит только у 25% пациентов с синдромом Крузона и наиболее часто изменены C2–C3. Kleiborg и соавторы сделали заключение, что сращение C5–C6 более характерно для синдрома Апера, а C2–C3 для синдрома Крузона, что помогает дифференцировать эти два заболевания [13]. Рентгенографическое исследование шейного отдела позвоночника является обязательным перед анестезиологическим пособием для этих пациентов. Schauerte и St-Aubin показали, что прогрессивный синостоз отмечается не только в черепных швах, но и в костях ног, рук, запястьях, шейном отделе позвоночника и предложили термин «прогрессирующий синостоз с синдактилией» как наиболее адекватно отражающий клиническую картину [24].
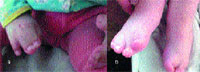 |
| Рисунок 3. Вид верхних (а) и нижних (б) конечностей больного С. с синдромом Апера |
Кожа. По некоторым данным, для синдрома Апера характерны элементы глазо-кожного альбинизма (светлые волосы и бледная окраска кожных покровов). Cohen и Kreiborg описали кожные проявления в 136 случаях синдрома [10]. Они обнаружили гипергидроз у всех пациентов. Также они описали акнеформные элементы, которые были особенно распространены на лице, груди, спине, руках. Помимо этого возможны проявления гипопигментации и гиперкератоза ладоней, западения кожи над крупными суставами конечностей. У некоторых пациентов имеется избыточная кожа складок лба.
Центральная нервная система (ЦНС). С синдромом связаны различные степени умственного дефицита, однако есть сообщения и о больных с нормальным интеллектом. Повреждения ЦНС в большинстве случаев могут быть причиной умственной отсталости. Возможно, проведение краниоэктомии на ранних этапах способствует нормальному умственному развитию. Patton и соавторы [20] проводили долгосрочное исследование 29 пациентов, из которых 14 имели нормальный или пограничный показатель интеллекта, у 9 отмечалась незначительнуая умственная отсталость (коэффициет интеллекта (IQ) 50–70), 4 были умеренно отсталыми (IQ 35–49) и 2 были выраженно отсталыми (IQ меньше 35). Ранняя краниоэктомия, казалось, не улучшала интеллектуальный статус. Шесть из 7 пациентов, окончивших школу, были приняты на работу или проходили дальнейшее обучение. Вопреки этим заключениям, Park и Powers, Cohen и Kreiborg утверждают, что многие из пациентов умственно отсталые [11, 19]. Они собрали информацию по 30 пациентам с патологией мозолистого тела, или структур лимба, или того и другого. Также у данных больных имелись и другие разнообразные нарушения. Авторы предположили, что эти аномалии могут быть причиной умственной отсталости. Прогрессирующая гидроцефалия встречалась редко, и часто ее не удавалось дифференцировать с непрогрессирующей вентрикуломегалией. Cinalli и соавторы обнаружили, что только 4 из 65 пациентов с синдромом Апера были шунтированы в связи с прогрессирующей гидроцефалией [8]. Renier и соавторы нашли уровень интеллекта 70 и больше у 50% детей из тех, кто имел декомпрессию черепа до 1 года, против 7,1% из тех, кто перенес оперативное лечение в позднем возрасте [22]. Патология corpus callosum (мозолистое тело) и размер желудочков мозга не коррелировались с заключительным показателем интеллекта, в отличие от патологии septum pellucidum (прозрачная перегородка). Качество окружающей среды и семейное окружение также определяют интеллектуальное развитие. Только 12,5% детей с данным синдромом имеют нормальные показатели интеллекта, по сравнению с 39,3% детей с нормальным внутрисемейным фоном.
Внутренние органы и системы. Для синдрома Апера характерны незначительные изменения со стороны внутренних органов. Патология со стороны сердечно-сосудистой системы (дефект межжелудочковой перегородки, несращенный Баталлов проток, стеноз легочной артерии, коарктация аорты, декстракардия, тетрада Фалло, эндрокардиальный фиброэластоз) отмечается у 10–20% больных. Аномалии мочеполовой системы (поликистоз почек, добавочные почечные лоханки, гидронефроз, стеноз шейки мочевого пузыря, двурогая матка, атрезия влагалища, увеличенные большие половые губы, клиторомегалия, крипторхизм) выявлены у 9,6%. Аномалии пищеварительной системы (пилоростеноз, атрезия пищевода, эктопия заднего прохода, частичная атрезия или недоразвитие желчного пузыря) обнаружены у 1,5%. Pelz и соавторы описали 18-месячную девочку, которая имела дистальный эзофагальный синдром в дополнение к типичным проявлениям синдрома Апера. Также в литературе упоминаются патологические изменения дыхательной системы — аномальные хрящи трахеи, трахеопищеводный свищ, легочная аплазия, отсутствие средней доли легкого, отсутствующие междолевые борозды [6, 11, 15].
Этиология синдрома Апера
За редкими исключениями синдром Апера вызывается одной из двух миссенс-мутаций гена FGFR2, вовлекающей две смежные аминокислоты: S252W и P253R, у 63% и 37% пациентов соответственно, по данным Wilkie и соавторов [29]. Park и соавторы исследовали корреляции фенотип/генотип у 36 больных с синдромом Апера [19]. Почти у всех, за исключением одного пациента, были найдены мутации S252W или P253R в гене FGFR2; частота составила 71 и 26% соответственно. Факт, что один пациент не имел мутации в этой области, дает основание предполагать наличие генетической гетерогенности синдрома Апера. Изучение 29 различных клинических проявлений продемонстрировало статистически несущественные различия между двумя подгруппами пациентов, имевших две основные мутации. Moloney и соавторы предоставили информацию относительно спектра мутаций и наследственного характера мутаций при синдроме Апера [16]. Их анализ 118 пациентов показал, что мутационный спектр при синдроме Апера узок. Мутация S252W наблюдалась у 74, а P253R — у 44 пациентов. Slaney и соавторы обнаружили отличия между клиническими проявлениями синдактилии и небной расщелины при двух основных мутациях гена FGFR2 при синдроме Апера [25]. Среди 70 пациентов с синдромом Апера 45 имели мутацию S252W и 25 — мутацию P253R. Синдактилия кистей и стоп была более серьезно выражена у пациентов с мутацией P253R. Напротив, расщелины неба оказались более характерны для пациентов с мутацией S252W. Различий в проявлении других патологий, связанных с синдромом Апера, найдено не было. Lajeunie и соавторы проводили скрининговое исследование 36 пациентов с синдромом Апера в целях обнаружения мутаций в гене FGFR2 [14]. Мутации были обнаружены во всех случаях. У 23 пациентов (64%) была обнаружена мутация ser252trp. У 12 пациентов (33%) была выявлена мутация pro253arg. Oldridge и соавторы проанализировали истории болезни 260 неродственных пациентов с синдромом Апера и нашли, что 258 имели миссенс-мутацию в экзоне 7 гена FGFR2, которая повреждала белок в линкерном районе между вторыми и третьими иммуноглобулиноподобными доменами [17]. Следовательно, генетическая причина возникновения синдрома Апера достаточно точно определена. Авторы установили, что 2 пациента имели вставки Alu-элемента в экзоне 9 или около него. Изучение фибробластов показало эктопическую экспрессию KGFR области FGFR2, которая была связана с выраженностью патологий конечностей. Эта корреляция оказалась первым генетическим свидетельством того, что аномальная экспрессия KGFR является причиной синдактилии при синдроме Апера. Основные миссенс-мутации в экзоне 7 (ser252trp и ser252phe) были выявлены у 258 и 172 пациентов соответственно. Von Gernet и соавторы проводили исследования относительно постхирургических проявлений в черепно-лицевой области у больных с различной степенью синдактилии [27]. У 21 пациента с синдромом Апера, из тех, кто подвергся хирургическому лечению краниофациальной области, лучшая клиническая картина была у больных с мутацией P253R, хотя они имели более серьезную форму синдактилии. Мутация P253R была определена у 6, а S252W — у 15 пациентов.
Диагностика и лечение
Удалось доказать, что больше чем 98% случаев вызваны определенными миссенс-мутациями, вовлекающими смежные аминокислоты (Ser252Trp, Ser252Phe или Pro253Arg) в экзоне 7 гена FGFR2, в связи с чем появилась возможность молекулярно-генетической диагностики синдрома Апера. Пока же этот метод не получил широкого распространения, основным способом диагностики является проведение компьютерной томографии (КТ) черепа. При помощи КТ выявляются такие характерные патологические изменения костей черепа, как коронарный синостоз, гипоплазия верхней челюсти, мелкие орбиты, изменения основания черепа и т. д. Наиболее наглядными являются данные, полученные при проведении КТ в формате ЗD. Магнитно-резонансная томография (МРТ) помогает оценить изменения мягких тканей черепа, связанные с костной патологией. Также для уточнения клинических проявлений синдрома Апера проводятся рентгенологические исследования костей верхних и нижних конечностей, целью которых является обнаружение различных форм костных синдактилий и изменений костей стоп и кистей. Помимо вышеперечисленных исследований, в диагностике степени выраженности фенотипических проявлений синдрома Апера и для прогноза развития заболевания важны данные психометрической оценки, исследования слуха, состояния дыхательных путей, а кроме того, заключения таких специалистов, как педиатр, клинический генетик, нейрохирург, ортодонт, отоларинголог, офтальмолог, невролог, психолог, логопед.
Хирургическое лечение включает в себя раннюю краниоэктомию коронарного шва и фронто-орбитальную репозицию для уменьшения проявлений дисморфизма и патологических изменений формы черепа. Операции по поводу синдрома Апера часто состоят из нескольких этапов, последний проводится в подростковом возрасте. Первый этап часто выполняется уже в 3 мес.
В последнее время стала широко использоваться новая техника краниофацильной дистракции с постепенным вытяжением кости. Этот метод приводит к хорошим косметическим результатам и снимает необходимость проведения костной пластики у пациентов в возрасте 6–11 лет. Помимо хирургического лечения патологии костей черепа, пациентам с синдактилией кистей и стоп проводится хирургическое лечение пальцев конечностей. Для формирования физиологического прикуса детям с синдромом Апера назначается ортодонтическое лечение.
Успехи в молекулярной генетике и неуклонное развитие клеточной биологии делают возможным понимание механизмов пороков развития у людей и их пренатальной диагностики. Определение фенотипа и генотипа и их корреляция очень важны для врача. Знание всех клинических проявлений того или иного синдрома позволяет хирургу выбрать правильную тактику ведения больных в пред- и послеоперационном периоде; помогает определить круг специалистов и исследований, необходимых для обследования пациентов. Практика показывает, что проблему лечения больных с синдромальными краниосиностозами нельзя решить при помощи изолированной работы краниофациальных хирургов. Как видно на примере синдрома Апера, синдромальные краниосиностозы сопровождаются не только деформацией костей черепа, но и патологическими изменениями как всего комплекса органов и тканей головы, так и костей скелета и внутренних органов. Для адекватного лечения больных с синдромальными формами краниосиностозов необходимо привлечение нейрохирургов, детских хирургов, педиатров, психологов, неврологов, окулистов, рентгенологов, отоларингологов, логопедов и генетиков. Наилучшие результаты достигаются при объединении усилий врачей всех перечисленных специальностей.
Литература
Д. Е. Колтунов, кандидат медицинских наук НПЦ медицинской помощи детям с пороками развития черепно-лицевой области и врожденными заболеваниями нервной системы, Москва



