Деградация белков
Деградация белков – означает разложение белков. Этот процесс происходит в организме непрерывно и компенсируется синтезом белков в мышцах. После тренировки при условии достаточного количества аминокислот, получаемых из питания, соотношение процессов разложения и синтеза в организме имеет позитивный характер.
Это означает преобладание синтеза белков и, как следствие, рост количества белков в организме и увеличение объема мышечной массы. Если организм не получает из продуктов питания достаточное количество белков, соотношение процессов синтеза и распада в организме имеет негативный характер. Это приводит к атрофии мышечной массы. Из всего этого следует необходимость ежедневного потребления достаточного количества белков, особенно при силовых тренировках и иной двигательной активности.
Протеолитическая деградация белков в клетке
Переставшие быть необходимыми, белки подвергаются протеолитической деградации.
Долгое время считали, что вышеупомянутому протеолизу подвергаются лишь белки, локализованные в цитоплазме, допускали возможность протеолиза ядерных белков. Сейчас ясно: система работает также в отношении белков, связанных с мембранами, секретируемых белков (для этого последние должны переместиться из эндоплазматического ретикулума в цитозоль путём обратного транспорта).
Система внутриклеточного протеолиза вовлечена в такие процессы как пролиферация клеток, развитие и дифференцировка, реакция на стресс и патогены, репарация ДНК. Нарушения этой сложной системы являются причиной многих заболеваний.
Стадии убиквитин-зависимого протеолиза
Деградация белка по убиквитиновому пути включает две основные стадии :
1. Ковалентное присоединение к подлежащему деградации белку полиубиквитиновой цепи.
2. Деградация белка 26S протеосомой.
Деградация белков, ассоциированных с мембраной
Процессирование белков, ассоциированных с мембраной, отличается от деградации цитоплазматических белков. Не вдаваясь в детали этого процесса, обсудим основные отличия :
1. Деградация осуществляется лизосомами.
2. Для таргетинга белка в лизосомы обычно достаточно моноубиквитинирования. В некоторых случаях формируется полиубиквитиновая цепь.
3. В случае формирования полиубиквитиновой цепи связывание происходит по 63 лизину.
Биологический смысл убиквитинирования
1. В строго определённый момент подвергать специфическому протеолизу огромное количество разнообразных белков.
2. Отменять деградацию, если белок всё ещё нужен клетке.
Биология клетки/Часть 1. Клетка как она есть/10/2


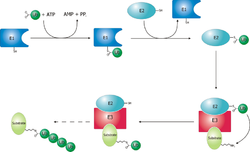
Белки не вечны. Они вступают в химические реакции, в результате которых «портятся», утрачивают работоспособность. Поэтому практически все белки в клетке и в организме регулярно заменяются. «Испорченные» белки внутри клеток метятся с помощью специального белка — убиквитина. Помеченные убиквитином белки поступают в специальные органоиды — протеасомы, внутри которых распадаются на отдельные аминокислоты.
Убиквити́н (от англ. ubiquitous — вездесущий) — небольшой консервативный белок, который у эукариот может присоединяться к другим белкам. Убиквитинирование — это посттрасляционное присоединение ферментами убиквитин-лигазами одного или, чаще, нескольких мономеров убиквитина с помощью ковалентной связи к боковым аминогруппам белка-мишени. Присоединение убиквитина влияет на внутриклеточную локализацию и функцию белков. Самым первым открытием роли убиквитина в клетке стало доказательство его участия в деградации белков. Белки, помеченных мультиубиквитиновыми цепями, расщепляются с помощью 26S- протеасомы. Однако впоследствии оказалось, что система убиквитина вовлечена и в такие важные процессы, как размножение и дифференцировка клеток, реакция на стресс и патогены, репарация ДНК.
В 2004 г Аарон Чехановер, Аврам Гершко и Ирвин Роуз были удостоены Нобелевской премии по химии «за открытие убиквитин-опосредованной деградации белка».
[1] Биомолекула.ru/ Вездесущий убиквитин
Маркировку белков, направляемых в протеасомы, осуществляет система убиквитинирования [ править ]
У млекопитающих до 90% клеточных белков (не только всех короткоживущих, но и большинства долгоживущих) подвергается гидролизу в полости протеасомы. Однако, прежде чем начнется этот процесс, она должна распознать объект протеолиза по какому-то признаку, ярлыку. Оказалось, маркировкой занимается специальная система ферментов (ее называют системой убиквитинирования). Маркером же служит цепочка не менее чем из четырех молекул белка убиквитина, состоящего из 76 аминокислотных остатков. Как образование цепочки через остаток лизина-48 в каждой молекуле, так и присоединение ее к белку-субстрату как раз и обслуживается системой ферментов. Эта система, включающая три типа ферментов (Е1, Е2 и Е3), высоко специфична и избирательна за счет того, что построена по принципу иерархического усложнения. Фермент Е1 (в клетке он только один) активирует молекулу убиквитина и передает ее одному из ферментов семейства Е2 (их называют конъюгирующими). Затем в каскад реакций вступает третий участник — представитель семейства Е3, лигаз, “сшивающих” ферментов. Он принимает убиквитин от Е2, соединяется с белком-субстратом и ковалентно пришивает к нему цепочку убиквитина. Если Е1 не имеет разновидностей, то семейство Е2 насчитывает 13 членов в клетке дрожжей Saccharomyces cerevisiae, а у млекопитающих — гораздо больше. В семействе Е3 сейчас известно около 100 разных лигаз, они-то и определяют в конечном счете высокую специфичность всей протеолитической системы.
Почему же цепочка убиквитина пришивается именно к тому белку, чья судьба предрешена? Оказывается, он уже несет признаки смерти — специфические сигналы, которые включают процесс деградации. Ими могут быть участки внутри белковой молекулы или на ее N-конце. Видимо, в определенных условиях они становятся доступными для узнавания ферментной системой, ответственной за маркировку. К основным сигналам для присоединения убиквитина могут быть отнесены следующие: • конформация N-терминальной области пептида, в частности наличие «дестабилизирующей» N-концевой или другой свободной -аминогруппы («N-концевое правило») или специфически расположенный лизин субстрата; • определенные короткие мотивы в последовательности аминокислотных остатков • нарушения вторичной и третичной структуры белка (неправильное свертывание полипептидной цепи); • повреждение боковых цепей остатков аминокислот, в том числе их окисление (например окисление остатков метионина); • избыточное гликозилирование белков и пептидов.
Некоторые N-концевые аминокислотные остатки (у эвкариот особенно часто Арг, Лиз, Лей, Фен, Асп) играют большую роль в определении жизни многих короткоживущих белков (в среднем они существуют от нескольких минут до трех часов), а также частично разрушенных белков или белков с измененной третичной структурой. В ряде случаев дестабилизирующие аминокислоты присоединяются к N-концу долгоживущих белков специфическими ферментами, после чего такие белки быстро разрушаются протеасомой.
На зависимость скорости деградации от природы N-концевых аминокислот (правило N-конца) первым обратил внимание наш бывший соотечественник А.Варшавский, он же ввел понятие “короткоживущие белки”.
Цепочка убиквитина способна присоединяться к белку-мишени и по сигналам, возникающим за счет некоторых вторичных модификаций (например, фосфорилирования) или соединения со вспомогательными белками.
Протеасома расщепляет белки на короткие пептиды,попадающие в цитоплазму [ править ]
Маркировка белка-субстрата (мишени) цепочкой убиквитина завершилась. Теперь ее узнает и связывается с ней одна или более субъединиц регулятора РА700. Этот процесс, как и последующее разворачивание субстрата, нуждается в энергии АТФ. Видимо, роль АТФазы выполняет тот же белок-регулятор. Развернутая, линейная молекула белка протягивается через регулятор, играющий роль рта протеасомы, и через открытое отверстие в a-кольце проникает в протеолитическую камеру. Здесь белок расщепляется на полипептиды длиной от 5 до 24 аминокислотных остатков. Они высвобождаются из протеасомы и в цитоплазме могут подвергнуться гидролизу до аминокислот протеазами (например, эндопептидазами). Часть этих полипептидов переносятся в лизосомы и затем перемещаются на поверхность клетки в комплексе с белками МНС, определяя ее антигенные свойства. Ненужная больше маркировочная цепочка из молекул убиквитина ликвидируется: изопептидазы разрывают ее на мономеры.
Работа протеасом играет важную роль в регуляции жизнедеятельности клетки [ править ]
Расщепление белков в протеасомах – главный механизм регуляции времени жизни короткоживущих белков. Видимо, протеасомы принимают участие и в процессах деградации белков и пептидов с аномальной структурой. Деградация короткоживущих регуляторных белков через убиквитин-протеасомный путь играет важную роль в основополагающих процессах жизнедеятельности клетки. К таким белкам, например, относятся циклины, циклин-зависимые киназы и их ингибитры, супрессоры опухолей, онкобелки, активаторы транскрипции и их ингибиторы и многие другие. Весьма детально изучена деградация циклинов – регуляторных белков, которые синтезируются и затем быстро разрушаются на различных фазах клеточного цикла, контролируя тем самым его прогрессию.
Итак, деградация белка в протеасомах:
Таким образом, внутриклеточный протеолиз — это не механический процесс деградации белков, а один из основных факторов, которые регулируют жизнедеятельность клетки.
Деградация белков-циклинов типа D

Выявлен молекулярный механизм разрушения одного из ключевых видов белков клеточного цикла — циклинов типа D. Этот механизм может лежать в основе отсутствия ответа некоторых опухолей на лечебное воздействие ингибиторными препаратами.
Белки, называемые циклинами D-типа (циклин D1, D2 и D3), — это ключевые компоненты ядерного комплекса, управляющего делением клетки. В своих работах в «Nature» Simoneschi с соавт. [1], Chaikovsky с соавт. [2] и Maiani с соавт. [3] отвечают на основные вопросы о деградации D-циклинов.
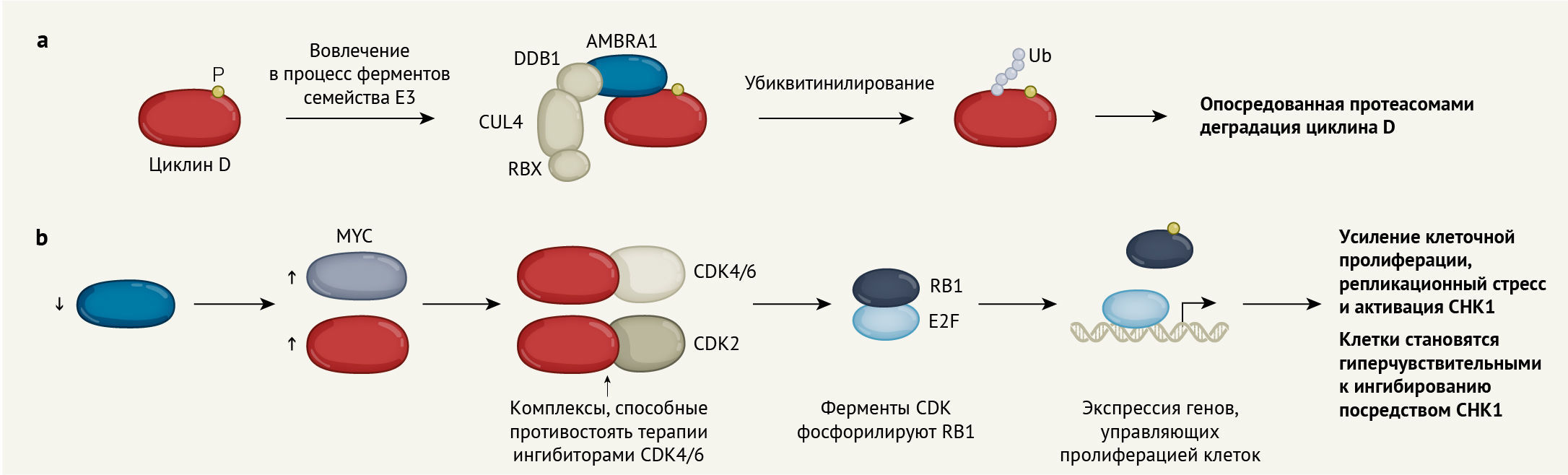
Во время деления клетки циклины D-типа связываются со своими ферментативными партнерами, называемыми циклин-зависимой киназой 4 (CDK4) и циклин-зависимой киназой 6 (CDK6), и активируют их. Эти киназы фосфорилируют белки-супрессоры опухолей RB1, RBL1 и RBL2, и это способствует клеточному делению (рис. 1). Неконтролируемая активация комплексов циклина D и киназ CDK4/6 является движущей силой в развитии многих типов рака [4].
В последнее время возрос интерес к метаболизму циклина D и CDK4/6, поскольку в клиническое применение вошли низкомолекулярные ингибиторы киназ CDK4/6. Результаты клинических испытаний этих веществ свидетельствуют об их способности увеличивать продолжительность жизни людей с раком груди. Ингибиторы CDK4/6 (палбоциклиб, рибоциклиб и абемациклиб) получили одобрение для лечения запущенных форм рака молочной железы. Более того, эти препараты задействованы в нескольких сотнях клинических испытаний, в ходе которых происходит поиск способов лечения многих различных типов рака [4, 5].
Циклины D-типа интенсивно изучаются с момента их открытия в 1990-е годы, но вопрос о том, как происходит их деградация во время клеточного цикла, остается спорным. Фосфорилирование карбоксильной концевой области D-циклинов запускает разрушение этих белков с помощью пути деградации, опосредованного убиквитин-протеасомной системой [6]. Система состоит из комплекса ферментов, включающего активатор убиквитина (E1), конъюгатор убиквитина (E2) и убиквитин-лигазу (E3); эти структуры связывают цепи нескольких молекул небольшого белка убиквитина с белками-мишенями посредством процесса, называемого убиквитинилированием. Эти убиквитинилированные белки затем подвергаются деградации в белковом комплексе, называемом протеасомой.
Самым большим семейством среди E3 являются cullin-RING лигазы (CRL). CRL состоят из cullin-белка, белка RING (который запускает E2), адаптивного белка и одного из многих различных белков субстрата-рецептора, ответственных за вовлечение целевого белка в комплекс E3 [7–9]. Некоторые субстратные рецепторы к E3, называемые CRL1, вовлечены в деградацию циклина D1, другие считаются мишенями воздействия для циклинов D2 и D3 для разрушения в протеасомах. Кроме того, как было выявлено, циклин D1 убиквитинилируется комплексом, запускаемым в анафазу, E3, который воздействует на несколько белков клеточного цикла [9]. В отличие от этих моделей функционирования, результаты других исследований [10] свидетельствуют, что на уровень и стабильность циклина D1 не влияет снижение содержания этих белков. Это указывает на то, что другие E3 регулируют деградацию циклина D1.
В статьях Simoneschi, Chaikovsky, Maiani с соавт. сообщается, что три циклина D-типа убиквитинилирутся и воздействуют на протеасомную деградацию с помощью подвида E3, называемого CRL4, с белком AMBRA1 в качестве рецептора субстрата. Уже было известно, что AMBRA1 играет ключевую роль в регуляции аутофагии — процесса, при котором клетки разрушают поврежденные органеллы или белковые структуры [11, 12]. Белок AMBRA1 также удалось идентифицировать как субстратный рецептор E3, в том числе и CRL4 [7, 13]. Проведя серию экспериментов с использованием методов клеточной, молекулярной биологии и генетики эмбриогенеза для получения сведений, одинаково применимых для биологии как мышей, так и человека, Simoneschi, Chaikovsky and Maiani с соавт. удалось продемонстрировать, что снижение концентрации AMBRA1 в нормальных и раковых клетках, а также в развивающихся эмбрионах мыши приводит к повышению содержания циклинов D-типа. Это приводит к большей степени фосфорилирования RB1 и большей пролиферации клеток, нежели чем это происходит в клетках с нормальным количеством AMBRA1.
Maiani с соавт. также продемонстрировали, что снижение содержания AMBRA1 приводит к повышению концентрации белка фактора транскрипции N-MYC. Этой же группой исследователей ранее было показано [14], что AMBRA1 регулирует стабильность и активность еще одного фактора транскрипции c-MYC. Белки семейства MYC могут повышать экспрессию циклинов D-типа и E-типа [15], тем самым ускоряя клеточный цикл.
Исходя из этих наблюдений, предполагается, что AMBRA1 может действовать как белок-супрессор опухоли. Действительно, у мышей с одной функциональной копией гена, кодирующего AMBRA1, чаще возникают опухоли легких, печени и почек, чем у нормальных мышей с двумя функциональными копиями этого гена [14]. Эти новые исследования предоставляют убедительные доказательства в поддержку данной гипотезы.
Авторы показывают, что при онкологическом заболевании у человека ген AMBRA1 мутирован. Как и ожидалось, учитывая то, что AMBRA1 способствует деградации циклина D1, авторы указывают, что уровень AMBRA1 в опухолевых тканях человека находился в обратной корреляционной зависимости от уровня циклина D1. Более того, низкий уровень AMBRA1 в опухолях оказался связан с плохим прогнозом для людей с онкологическими процессами. Экспериментальная инактивация AMBRA1 либо в опухолевых клетках человека, либо в клетках мыши, выращенных с помощью методов генной инженерии с мутациями, приводящими к развитию рака, увеличила опухолевый потенциал клеток, что было оценено после введения инъекции мышам. Кроме того, удаление гена AMBRA1 стимулировало образование опухоли (что было продемонстрировано на мышиной модели рака легкого), вызванной мутантной версией гена Kras, и у этих AMBRA1-дефицитных опухолей обнаружился более высокий уровень D-циклинов. Данные всех трех исследований свидетельствуют о том, что в основном AMBRA1 сдерживает пролиферацию клеток, не давая D-циклинам достигать высоких концентраций.
Кроме того, Chaikovsky, Simoneschi с соавт. демонстрируют, что исчезновение AMBRA1 и соответствующее повышение концентрации D-циклинов приводит к снижению чувствительности опухолевых клеток человека к ингибиторам CDK4/6. Интересно, что эти же авторы указывают, что в клетках со сниженной концентрацией AMBRA1, а не в комплексе с CDK4/6, циклин D1 также образует каталитически активный комплекс с циклин-зависимым ферментом киназой CDK2. Также у этих комплексов отсутствует чувствительность к ингибиторам CDK4/6.
Maiani с соавт. также указывают, что исчезновение AMBRA1 и, как следствие, повышение содержания D-циклинов (и, возможно, также других белков, таких как c-MYC), вызывает повреждение ДНК и репликационный стресс, который сопровождается активацией фермента киназы CHK1. Важно отметить, что Maiani с соавт. сообщают о том, что раковые клетки со сниженной концентрацией AMBRA1 гиперчувствительны к лечению ингибиторами CHK1, что предполагает потенциальную возможность терапевтического воздействия на опухоли, у которых обнаруживается дефицит AMBRA1.
Эти удивительные результаты поднимают несколько важных вопросов. Например, может ли снижение уровня AMBRA1 лежать в основе существующей или приобретенной устойчивости опухолей человека к ингибиторам CDK4/6? Является ли повышение содержания D-циклинов, которое наблюдается при исчезновении AMBRA1, единственным фактором, ответственным за развитие устойчивости к ингибиторам CDK4/6? Анализ клинических испытаний на людях с раком груди [16–19] не выявил корреляции между наличием добавочных копий гена, кодирующего циклин D1, или уровнем матричной РНК циклина-D1, или белка в опухолях и реакцией пациента на ингибиторы CDK4/6. Действительно, Chaikovsky с соавт. обнаружили, что раковые клетки человека, у которых более выражена экспрессия D-циклинов по сравнению с нормой, не обладают всеми характеристиками устойчивости к ингибиторам, как при почти полном исчезновении AMBRA1. Возможно, другие белки, регулируемые AMBRA1, такие как c-MYC, которые могут активировать белок циклин E и комплексы циклин E-CDK2, влияют на устойчивость к лечению.
Наблюдение Chaikovsky с соавт. и Simoneschi с соавт. того, как образуются комплексы циклин D – CDK2, устойчивые к препаратам-ингибиторам CDK4/6, в клетках, в которых снижено содержание AMBRA1, является невероятно занимательным для молекулярной онкологии. Как было обнаружено ранее, такие «атипичные» комплексы лежат в основе приобретенной устойчивости к ингибированию CDK4/6 [20]. Итак, можно предположить, что снижение концентрации AMBRA1 каким-то образом способствует образованию этих комплексов циклина D с CDK2, и это наряду с повышением концентрации циклина D приводит к устойчивости к ингибиторам CDK4/6. Особенно интересная перспектива, проистекающая из работы Maiani с соавт., заключается в том, что ингибиторы CHK1 можно использовать для лечения опухолей, устойчивых к ингибиторам CDK4/6, в которых крайне мало содержание AMBRA1.
Необходимы дальнейшие исследования роли AMBRA1 в развитии рака у человека. Опосредуется ли супрессивная функция AMBRA1 на опухоль преимущественно циклином D1 или c-MYC, либо же она также опосредована иными соединениями? Опухолевые клетки, которые больше не продуцируют RB1, не требовательны к наличию D-циклинов для клеточного цикла [4], поэтому, как можно предположить, исчезновение AMBRA1 будет наблюдаться в опухолях, которые продуцируют RB1, в случае, если D-циклины являются основной молекулярной мишенью AMBRA1. Также еще предстоит определить, является ли исчезновение AMBRA1 в опухолях человека взаимоисключающим параметром в связи с наличием мутаций, затрагивающих C-концы D-циклинов, которые, как предполагают, могут сделать D-циклины устойчивыми к деградации, опосредованной AMBRA1. Другой нерешенный вопрос заключается в том, почему (по данным Chaikovsky с соавт.), существует корреляционная зависимость между низкой степенью экспрессии AMBRA1 и высокими концентрациями циклина D1. Также неясно, почему эта низкая степень экспрессии AMBRA1 связана с плохой выживаемостью при опухолях легких, в которых присутствуют определенные типы генетических изменений, например, мутации Kras. Этот эффект не наблюдается для опухолей легких, в которых вместо этой мутации были мутантные версии гена, кодирующего белок EGFR, или Kras дикого типа.
Независимо от ответов на эти вопросы, впечатляющие исследования Chaikovsky с соавт., Maiani с соавт. and Simoneschi с соавт. вносят свой важный вклад в понимание механизмов, управляющих клеточным циклом.
Что такое деградация белка
ПРОТЕАСОМА:
РАЗРУШЕНИЕ ВО ИМЯ СОЗИДАНИЯ
Е.Б. Абрамова, В.Л. Карпов
Известно, что в живой клетке содержатся множество разных белков. Одни из них существуют довольно долго, другие живут от нескольких минут до 2—3 ч. Последние синтезируются только в определенный момент жизни клетки, в ответ на некоторые внутренние и внешние импульсы. Когда потребность в таком белке отпадает, специальные факторы сигнализируют о том, что его синтез должен быть остановлен. Действуют они или на этапе считывания матричных РНК (мРНК) с ДНК (факторы транскрипции), или на стадии синтеза белка с уже имеющихся РНК-матриц. Однако, если некий белок существует, но в его функционировании клетка больше не нуждается, должен быть и механизм, обеспечивающий остановку его работы. Такой механизм давно и весьма детально исследован для белков-ферментов. Чтобы “вывести из строя” фермент, его активность обратимо подавляется (ингибируется) веществами белковой или небелковой природы. Однако клетка на протяжении своей жизни синтезирует и множество белков, не обладающих ферментативной активностью, но тем не менее “исполняющих” разнообразные роли. Такие белки не только прекращают свою работу в определенный момент, но и существуют очень недолго (потому и называются короткоживущими) по сравнению со временем жизни клетки.
Очевидно, что каким-то образом должны удаляться и “сделавшие свое дело” белки, иначе они переполнят клетку и разрушат ее. Может быть, она справляется с ними посредством хорошо изученного протеолиза (распада белков под действием специальных ферментов)? Но тогда неясно, почему не повреждаются при этом структурные компоненты клетки и еще нужные ей белки. Протеолиз в лизосомах — процесс неспецифический, в этих образованных мембраной “мешочках” с набором ферментов гидролаз молекулы белков расщепляются до аминокислот, идущих затем в обменные процессы клетки (здесь же гидролизуются нуклеиновые кислоты и полисахариды). У высших эвкариот лизосомы разрушают только белки, связанные с мембранами, а также чужеродные, захваченные во время эндоцитоза (например, вирусные или бактериальные).
К началу 80-х стало ясно, что эффективная регуляция не только количества, но и функции многих белков зависит и от процессов, связанных с деградацией.
В это же время в клетке был обнаружен высокомолекулярный белковый комплекс, который работал в определенных условиях как несколько протеолитических ферментов [1]. Однако, что очень важно, свою разрушающую активность комплекс проявлял только в явно нефизиологических условиях под действием изменяющих его структуру веществ. Он был найден в клетках как самых примитивных, так и высших эвкариот, причем и в ядре, и в цитоплазме. Это свидетельствовало об абсолютной необходимости комплекса для нормальной жизнедеятельности клетки.
Его начали интенсивно изучать во многих лабораториях мира, и даже возникло множество названий, из которых сейчас чаще всего используется то, что дали К.Танака и А.Голдберг, — протеасома, т.е. частица (сома) с протеолитической функцией [2]. Надо сказать, что протеасомой названы две частицы разной сложности строения. Они отличаются также молекулярной массой и коэффициентом седиментации (его выражают в единицах Сведберга и обозначают буквой S при числе).
В 90-х годах выяснилось, что первоначально выделенный комплекс с молекулярной массой около 700 кДа и коэффициентом седиментации 20S в качестве протеолитического ядра входит в состав еще более сложной частицы. Первую стали называть 20S протеасомой, вторую — 26S протеасомой.
Компьютерная томография двух форм протеасомы [8].
Сборка 20S протеасомы.
Схематическое строение 26S протеасомы и протеолитических камер.
Регулятор РА700 присоединяется к обоим концам протеолитического ядра-цилиндра (эта реакция также нуждается в энергии, ее источником служит АТФ) и образует зеркально симметричную структуру, похожую на гантели.
Каталитическое ядро (т.е. 20S протеасома), связанное с двумя регуляторами РА700, и есть 26S протеасома, ее молекулярная масса составляет более 2.5 МДа. Этот комплекс обозначают и как РА700-20S-РА700.
Заметим, что с 20S протеасомой могут связаться и оба регулятора сразу, тогда возникает гибридная форма РА700-20S-РА28. Как будут распределены разные формы и в каком количестве понадобятся клетке, зависит от ее молекулярных потребностей в определенное время ее жизни и в конкретных обстоятельствах. Об этом мы еще расскажем, а сейчас вернемся к 26S протеасоме, вернее, к ее субстратам — белкам.
Итак, молекулярная машина собрана и готова работать — разрушать отработавшие белки, чтобы клетка не только не заполнилась ими до отказа, но и использовала белковые осколки для своих целей.
У млекопитающих до 90% клеточных белков (не только всех короткоживущих, но и большинства долгоживущих) подвергается гидролизу в полости протеасомы. Однако, прежде чем начнется этот процесс, она должна распознать объект протеолиза по какому-то признаку, ярлыку. Оказалось, маркировкой занимается специальная система
Различные формы протеасомы и ее активаторов.
ферментов (ее называют системой убиквитинирования). Маркером же служит цепочка не менее чем из четырех молекул белка убиквитина, состоящего из 76 аминокислотных остатков. Как образование цепочки через остаток лизина-48 в каждой молекуле, так и присоединение ее к белку-субстрату как раз и обслуживается системой ферментов.
Эта система, включающая три типа ферментов (Е1, Е2 и Е3), высоко специфична и избирательна за счет того, что построена по принципу иерархического усложнения. Фермент Е1 (в клетке он только один) активирует молекулу убиквитина и передает ее одному из ферментов семейства Е2 (их называют конъюгирующими). Затем в каскад реакций вступает третий участник — представитель семейства Е3, лигаз, “сшивающих” ферментов. Он принимает убиквитин от Е2, соединяется с белком-субстратом и ковалентно пришивает к нему цепочку убиквитина.
Маркировка белка-субстрата (мишени) цепочкой убиквитина завершилась. Теперь ее узнает и связывается с ней одна или более субъединиц регулятора РА700. Этот процесс, как и последующее разворачивание субстрата, нуждается в энергии, которую поставляют, видимо,
Описывая каскад реакций по маркировке субстрата, мы, чтобы не разбивать ход событий, намеренно кое-что опустили. Не сказали, почему цепочка убиквитина пришивается именно к тому белку, чья судьба предрешена. Оказывается, он уже несет признаки смерти — специфические сигналы, которые включают процесс деградации. Ими могут быть участки внутри белковой молекулы или на ее N-конце. Видимо, в определенных условиях они становятся доступными для узнавания ферментной системой, ответственной за маркировку.
Некоторые N-концевые аминокислотные остатки (у эвкариот, особенно Арг, Лиз, Лей, Фен, Асп) играют большую роль в определении жизни многих короткоживущих белков (в среднем они существуют от нескольких минут до трех часов), а также частично разрушенных или с измененной третичной структурой. На зависимость скорости деградации от природы N-концевых аминокислот (правило N-конца) первым обратил внимание наш бывший соотечественник А.Варшавский, он же ввел понятие “короткоживущие белки” [3]. В ряде случаев дестабилизирующие аминокислоты присоединяются к N-концу долгоживущих белков специфическими ферментами, после чего такие белки быстро разрушаются протеасомой.
Различные формы протеасомы и ее активаторов.
Схема протеасомной деградации белков.
Вверху — образование убиквитиновой цепочки;
внизу — гидролиз субстрата протеасомой до пептидов и свободного убиквитина.
Цепочка убиквитина способна присоединяться к белку-мишени и по сигналам, возникающим за счет некоторых вторичных модификаций (например, фосфорилирования) или соединения со вспомогательными белками.
В общих чертах мы рассмотрели всю схему (именно схему, потому очень многие подробности остались за ее пределами) строения и сборки протеасомы, систему маркировки белка, который будет ею разрушен до полипептидов. За счет этого протеасома регулирует время жизни важнейших белков, удаляет из нее чужеродные и аномальные, поставляет образовавшиеся в результате гидролиза полипептиды в качестве антигенов, способных сообщать иммунной системе о неполадках в клетке. Таким образом, внутриклеточный протеолиз — это не механический процесс деградации белков, а один из основных факторов, которые регулируют жизнедеятельность клетки.
Из разнообразных процессов, в которых участвует протеасома (табл.), рассмотрим лишь некоторые.
Участница множества процессов
Отметим прежде всего, что при изучении роли протеасомной деградации в различных клеточных процессах весьма часто использовались ингибиторы активности протеасомы. Они легко проникают в клетку и избирательно подавляют данный путь протеолиза [4].
Регуляция клеточного цикла. Как известно, клеточный цикл состоит из нескольких фаз, а их последовательная смена регулируется белками циклинами. Поскольку на каждой фазе действует собственный регулятор, жизнь его должна быть короткой. Это и обеспечивает 26S протеасома. Оказалось, что одни циклины в качестве метки для узнавания содержат участки, обогащенные пролином, глутаминовой кислотой, серином и треонином, а ряд других — консервативный фрагмент из девяти аминокислот, который обычно располагается на расстоянии около 40 аминокислотных остатков от N-конца. Узнанный по той или иной метке регуляторный белок сшивается, как уже было сказано, с убиквитиновой цепочкой своим собственным ферментом из семейства Е3 и разрушается 26S протеасомой. Ясно, что сбой в ее работе вызовет остановку клеточного цикла на той или иной фазе.
Злокачественное перерождение клетки. В нормальной здоровой клетке белки, регулирующие скорость транскрипции, во многих случаях определяют ее дальнейшую судьбу — станет ли работать и делиться с запрограммированной для нее скоростью, пойдет ли по пути неконтролируемого злокачественного роста, будет ли разрушена как представляющая опасность. Поэтому такие регуляторные белки в зависимости от обстоятельств могут быть или онкобелками, или же, наоборот, онкосупрессорами. Пример подобных превращений — белок р53, уровнем которого поддерживается в здоровой клетке соотношение между процессами роста и апоптоза. Но все меняется, например, при заражении человека вирусом папилломы: вирусный белок E6 находит белок р53 и сигнализирует строго определенному ферменту из семейства Е3 о необходимости присоединить к р53 убиквитиновую цепочку. Фермент выполняет свою функцию, и р53 становится субстратом для протеасомной деградации. В результате его ускоренного разрушения клетка идет по пути злокачественного перерождения.
Транскрипция. Мы говорили, что под действием 26S протеасомы расщепляются белковые молекулы. Однако выяснилось, что протеолизу может подвергаться даже отдельная субъединица в сложном белковом комплексе. Представить себе механизм взаимодействия с ним 26S частицы довольно трудно. Тем не менее частичный протеолиз осуществляется в процессе двухступенчатой активации ядерного фактора (его обозначают NF-kB), который контролирует транскрипцию. Сначала в результате гидролиза протеасомой образуется из своего предшественника субъединица p50 этого фактора. Затем она, еще одна субъединица — р65 — и белок-ингибитор IkB объединяются в неактивный комплекс. Транскрипция становится возможной после деградации 26S протеасомой ингибитора IkB, перед этим фосфорилированного в двух местах.
Иммунная система. Участие протеасомы в иммунном ответе клетки — один из наиболее активно изучаемых аспектов работы 26S частицы. Она, гидролизуя аномальные или чужеродные белки до полипептидов, поставляет некоторые из них (обычно длиной от восьми до 11 аминокислот) в качестве антигенов [5]. Такие полипептиды соединяются в цитозоле с определенным транспортным белком и переносятся в эндоплазматический ретикулум, где взаимодействуют с молекулами белков класса I главного комплекса гистосовместимости и выносятся на поверхность клетки. Вновь появившиеся антигены иммунная система обнаруживает с помощью цитотоксических T-лимфоцитов и разрушает клетки, в которых продуцируются вирусные или другие необычные для них белки.
Образование иммуногенных пептидов за счет расщепления вирусного белка протеасомой и их вынос на поверхность клетки в качестве антигенов. Весь путь пептидов показан цветом. Клетка с такими антигенами на поверхности узнается цитотоксическими лимфоцитами и разрушается ими. ГКГС1 — главный комплекс гистосовместимости, молекулы класса 1.
Мы упоминали, что существуют три формы протеасомы (в зависимости от того, с каким регулятором связано ее ядро): РА700-20S-РА700, т.е. 26S форма, РА28-20S-РА28 и гибридная РА700-20S-РА28. Предполагают, что вторая форма способна гидролизовать длинные пептиды — первоначальный продукт расщепления белков 26S протеасомой — до коротких. Именно они и выносятся на поверхность клетки в качестве антигенов. Что касается гибридной формы, то один ее регулятор (РА700), видимо, узнает белок, несущий убиквитиновую цепочку, а второй (РА28) заставляет каталитическое ядро гидролизовать белковую молекулу до более коротких иммуногенных пептидов. К настоящему времени установлено, что содержание и внутриклеточное распределение форм протеасомы связано с действием g-интерферона и что в цитоплазме одновременно с уменьшением количества 26S протеасомы накапливается гибридная форма.
Термозащита. При нагревании, как известно, белки утрачивают нормальную конфигурацию и перестают выполнять свои функции. Чтобы защитить клетку от столь пагубных последствий, в ней синтезируются белки теплового шока, или шапероны. Они исправляют нарушенную высокой температурой форму белковых цепей. В каком качестве участвует в этом процессе протеасома, пока не совсем ясно. Однако установлено, что, если подавить ее активность, в клетке накапливаются поврежденные белки, но в то же время стимулируется синтез мРНК шаперонов. Повышается также и уровень трегалозы (молекулы-термозащитницы) и термостабильность клеток в целом. Следовательно, полагают, ингибиторы протеасомы могут найти применение в медицине в качестве терапевтических средств при шоковых воздействиях высокими температурами.
Эту отнюдь не простую связь изучают в основном с помощью ингибиторов активности протеасомы, и интенсивнее всего в исследованиях онкогенеза. Оказалось, что, снижая уровень активности упомянутого уже ядерного фактора транскрипции (NF-kB), такие ингибиторы могут запускать апоптоз трансформированных клеток, предотвращать ангиогенез (разрастание кровеносных капилляров в раковой ткани) и метастазирование in vivo. Это результат того, что снижение каталитической активности протеасомы приводит к накоплению ингибиторов роста клетки и проапоптозных белков. Не разрушенные ингибированной протеасомой короткоживущие белки, такие как р53 и р27, тоже способны запустить в клетке каскад биохимических и морфологических событий, ведущих к апоптозу. Однако ингибиторы протеасомы могут и предотвращать его, например в первичных клеточных культурах, если он вызван какими-то другими стимулами.
Как видно из приведенных примеров, ингибиторы протеасомной деградации служат молекулярным инструментом, с помощью которого удается раскрыть связь между клеточной смертью и работой протеасомы. Но не менее важно и другое: если к апоптозу злокачественных клеток приводят ингибиторы, возможно, они могут послужить медицине как противораковые средства.
Белки-субстраты и болезни
Деградация белков протеасомой — процесс с тонкой настройкой, поэтому сбои в нем, нарушающие равновесие между пролиферацией и апоптозом, служат причиной разных болезней как врожденных, так и приобретенных. Условно их можно разделить на две группы: заболевания, обусловленные тем, что деградационная система не работает, и болезни, которые возникают из-за усиления ее функции. Первые — это результат стабилизации субстратов, быстро разрушаемых в норме, вторые, наоборот, — аномально быстрого распада белков-мишеней.
Мы говорили, что клеточный белок р53 при заражении онкогенным штаммом вируса папилломы человека соединяется с вирусным онкобелком Е6 и становится субстратом для ускоренной деградации протеасомой. В результате лишенная защиты клетка — мишень вирусной атаки — превращается в раковую. Действительно, при некоторых формах карциномы уровень клеточного р53 бывает резко снижен. Предполагают, что таким способом вирус может неконтролируемо размножаться в клетке, и она становится злокачественной. Агрессивные формы рака прямой кишки и молочной железы исследователи тоже связывают с работой протеасомной системы, хотя механизм образования трансформированных клеток здесь другой.
Видимо, некоторые врожденные заболевания человека обусловлены генетическими изменениями или в белках-субстратах, или в ферментах, отвечающих за сшивку с убиквитином. Фиброкистоз, например (его признаки — хроническая закупорка и инфекция дыхательных путей, нарушение пищеварения), связан с мутацией,
приводящей к преждевременному гидролизу протеасомой мембранного белка, который регулирует транспорт ионов хлора через мембрану эпителиальных клеток. При синдроме Ангельмана (задержка умственного развития) обнаружены делеция части Х-хромосомы и поврежденные молекулы фермента из семейства Е3. Редкая наследственная форма гипертензии, или синдром Лиддла, обусловлена делецией в генах субъединиц белка, который формирует натриевые каналы в клетках эпителия. В итоге меняется скорость гидролиза субъединиц протеасомой и, как следствие, — баланс между ионами калия и натрия (т.е. нарушается солевой гомеостаз).
Целый ряд известных аутоиммунных заболеваний может быть вызван неправильным разрезанием белка протеасомой. Она ведь гидролизует и собственные нормальные клеточные белки, и чужеродные. Но из образующихся пептидов только чужие становятся антигенами, именно по ним цитотоксические лимфоциты узнают зараженную клетку и уничтожают ее. Свои же пептиды не вызывают Т-клеточного ответа. Однако, если по какой-либо причине протеасома неверно расщепляет субстрат, то и из собственных белков могут возникать нетипичные пептиды, которые распознаются Т-лимфоцитами как чужеродные. И тогда клетки организма, ничем не зараженные, становятся мишенью для атаки. По-видимому, с протеасомной системой связаны многие иммунные заболевания и воспалительные реакции, но конкретные механизмы их возникновения отнюдь не одинаковы.
Интересно, что вирусы могут уйти из-под контроля иммунной системы, причем разными способами. Вирус гепатита В, например, достигает этого за счет взаимодействия собственного белка Х с субъединицами 20S протеасомы и регулятора РА28. В такой комбинации протеолиз подавляется и белок Х остается целым. А раз нет пептидов-антигенов, иммунная система не воспринимает зараженную клетку. Цитомегаловирус человека не предохраняет свои белки от гидролиза, а способствует разрушению белковых молекул главного комплекса гистосовместимости, которые отвечают за транспорт: пептиды-антигены не попадают на поверхность зараженной клетки, и та выживает. Другие вирусные белки взаимодействуют с протеасомными АТФазами, увеличивая скорость гидролиза внутриклеточных белков и приводя к усиленному размножению вируса.
Установлено, что в дистрофии скелетных мышц, обусловленной голоданием, сепсисом и денервацией, виноват усиленный гидролиз мышечных белков протеасомой. Но активирующие ее внеклеточные стимулы и сигнальные пути пока не ясны.
Предполагают, что накопление окисленных белков при старении связано с нарушениями протеасомной деградации. И это не лишено основания.
* * * Прошло 20 лет с открытия специфического внутриклеточного протеолиза. За это время в мире сложилось несколько научных центров по изучению протеасомы, ее генов и регуляции их считывания. Надо сказать, что в лаборатории структуры и функции хроматина Института молекулярной биологии им. В.А.Энгельгардта РАН обнаружена новая сигнальная цепь, которая контролирует зависимый от АТФ гидролиз белков протеасомой [6, 7]. Оказалось, у пекарских дрожжей ( Saccharomyces cerevisiae ) короткоживущий белок Rpn4 служит фактором транскрипции, регулирующим работу более 500 генов, которые кодируют большинство субъединиц протеасомы и ферменты, участвующие в протеасомной деградации белков. Таким образом, впервые показано, что расщепление белков контролируется и на уровне транскрипции генов.
Сейчас уже никто не сомневается, что без внутриклеточного протеолиза, который специфически “выводит из игры” в нужное время и в нужном месте важнейшие белковые компоненты, клетка не может обойтись. Сделано очень много, а предстоит еще больше, потому что работа протеолитической частицы теснейшим образом смыкается с патогенезом многих заболеваний. А значит, и с возможностью изменять их течение.
Работа поддерживается Российским фондом фундаментальных исследований. Проект 03-04-49127.
1. Wilk S., Orlowski M. // J. Neurochem. 1983. V.40. P.842—849.
2. Arrigo A.-P., Tanaka K., Goldberg A.L., Welch W.J. // Nature. 1988. V.331. P.192—194.
3. Bachmair A., Finley D., Varshavsky A. // Science. 1986. V.234. P.179—186.
4. Rock K.L., Gramm C., Rothstein L. et al. // Cell. 1994. V.78. P.761—771.
6. Mannhaupt G., Schall R., Karpov V. et al. // FEBS Lett. 1999. V.450. P.27—34.
7. Капранов А.В., Курятова А.В., Преображенская О.И., Карпов В.Л. // Мол. биология. 2001. №35. С.356—364.
8. Baumaister W., Walz J., Zuhl F., Seemuller E. // Cell. 1998. V.92. P.367—380.



