Орфанные заболевания. Редкие и опасные

Обсуждаемой новостью стало, что часть средств, вырученных от увеличения ставки НДФЛ, планируется направить в целевой фонд на лечение детей с орфанными заболеваниями. Подробнее об орфанных заболеваниях и как они изучаются в России в нашем материале.
Что такое орфанные заболевания?
Редкость заболевания, с одной стороны, плюс, так как немногие люди сталкиваются с ними, но для носителей заболевания – существенный минус. Недостаточная изученность болезни затрудняет постановку диагноза, а в следствии, и оказание своевременного лечения.
Минздрав России 27 февраля 2020 года опубликовал перечень орфанных заболеваний, куда входят 258 болезней, включая различные новообразования, заболевания кровообращения, эндокринные нарушения, врождённые аномалии, психические расстройства и другие.
Лечение орфанных заболеваний дорогостоящее. Например, для лечения детей со спинальной мышечной атрофией применяют препарат стоимостью около восьми миллионов рублей за одну инъекцию.
В России существуют программы по лечению орфанных заболеваний на региональном и федеральном уровнях, но многие родители вынуждены обращаться к помощи благотворительных фондов, самостоятельному поиску средств на лечение детей через Интернет. В такой ситуации необходима системная поддержка на государственном уровне.
Как изучаются орфанные заболевания?
По информации Минобрнауки, в России существует всего несколько научных центров и лабораторий, где изучаются орфанные заболевания и генные патологии, в том числе Лаборатория «Молекулярная медицина и генетика человека» СВФУ, Лаборатория терапии орфанных заболеваний МФТИ, Медико-генетический научный центр имени академика Н.П. Бочкова. Эти лаборатории занимаются изучением причин и механизмов развития орфанных и мультифакториальных заболеваний, разрабатывают способы диагностики и профилактики этих заболеваний.
Особое место в работе лаборатории «Молекулярная медицина и генетика человека» СВФУ занимает исследование генетических заболеваний, характерных для представителей якутского этноса, в связи с чем она работает в тесном сотрудничестве с Национальным центром медицины Республики Саха (Якутия). Изучаются: SOPH-синдром, приводящий к низкорослости с атрофией зрительных нервов, 3-М синдром, так называемый «якутский синдром низкорослости», врожденная глухота первого А-типа, наследственная метгемоглобинемия и синдром мукополисахаридоз-плюс и другие заболевания. Все разработки СВФУ – новые методы диагностики и лечения – сразу внедряются в практическое здравоохранение.
В лаборатории терапии орфанных заболеваний МФТИ при совместной работе с Национальным медицинским исследовательским центром эндокринологии Минздрава РФ специалисты работают над созданием терапии для врожденной дисфункции коры надпочечников – группы заболеваний, обусловленных нарушением стероидогенеза в результате дефицита одного из ферментов, участвующих в синтезе кортизола. Предлагаемая лабораторией стратегия терапии основывается на интеграции нормальной копии гена в геном стволовых клеток коры надпочечников для обеспечения пожизненного терапевтического эффекта, что станет альтернативой применяющейся гормональной терапии. Кроме того, в лаборатории геномной инженерии МФТИ по заказу компании «Артема» ведется разработка генной терапии колбочковой дистрофии сетчатки глаза с измененным палочковым ответом. В случае успешного тестирования, терапия позволит остановить потерю зрения у людей с генной мутацией, а также поможет восстановить зрение уже ослепшим из-за наследственной патологии пациентам.
Сотрудники Медико-генетического научного центра имени академика Н.П. Бочкова разрабатывают уникальные методы эффективной диагностики наследственных болезней, а также новые методы лечения, лекарственные средства, способы диагностики и индивидуализации терапии онкологических заболеваний. Для каждого конкретного пациента разрабатывается новый, уникальный метод функционального анализа. В 2019 году специалисты МГНЦ осуществили консультативный прием 11 тысяч пациентов и провели свыше 13 тысяч генетических исследований в рамках госзадания. Центр является первым в Европе по количеству диагностируемых наследственных заболеваний и единственной организацией в России, выполняющей высокотехнологичные генетические тесты на бюджетной основе в таком объеме.
«Уровень развития медицинской генетики в нашей стране соответствует общемировому. В России сегодня есть возможности применения всех современных подходов диагностики и профилактики наследственных болезней. Развиваются полногеномные исследования, анализ хромосом, подходы профилактики наследственной и врожденной патологии. При этом технологии, которыми владеют врачи в Российской Федерации, не отличаются от технологий, применяющихся в развитых странах. Сегодня у нас меняется парадигма: мы говорим уже не о симптоматическом, а о патогенетическом лечении, о развитии таких современных технологий, как генотерапия. Мы подошли очень близко к тому, чтобы получить лекарственные препараты для лечения ряда наследственных заболеваний. Совместная работа с международным консорциумом Orphanet позволит сделать дальнейшие шаги в этом направлении», – сообщил Сергей Куцев, директор ФГБНУ «МГНЦ», главный внештатный специалист по медицинской генетике Минздрава России.
«Оптимисты». Как живут люди с орфанными заболеваниями в России?
Ника Воюцкая
Павлику, сыну Снежаны Митиной, было всего три года, когда ему поставили диагноз мукополисахаридоз II типа, или синдром Хантера. «Мы только открыли дверь, а врач уже говорит: „Какой прекрасный Хантер!“ — рассказывает Снежана теперь. — С первой частью я согласилась, а вот кто такой Хантер, мне тогда было неизвестно».
В детском саду мальчик был самым высоким в своей группе, читал стихи про бычка, который идет и качается. Поверить, что совсем скоро он перестанет ходить и говорить, казалось невозможным.
Но в четыре из-за гиперактивности Павлика исключат из детского сада, в шесть — из детского сада для инвалидов: перестанет усваивать программу. У него изменятся внешность и характер.
Терапии, способной корректировать часть симптомов, вместе с другими родителями Снежане придется добиваться самой. Элапразу — единственный препарат, способный помочь ее сыну, зарегистрируют в России не сразу и именно ее трудами.
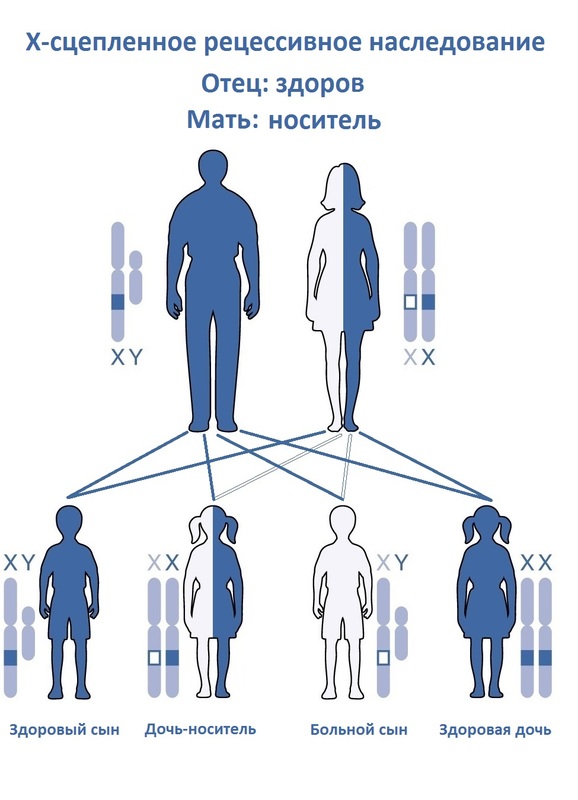
Синдром Хантера — это одна из форм мукополисахаридоза, генетическое заболевание, возникающее в результате дефицита ряда ферментов, расщепляющих продукты обмена. Этот дефицит приводит к накоплению белково‑углеводных комплексов и жиров в клетках. В результате чего происходит «самоотравление» организма и поражаются органы.
В России редким, или орфанным, считается заболевание, которое встречается у 1 человека из 10 тысяч или реже. Именно из-за редкости такие болезни сложно диагностировать и лечить. Тот синдром, что у Паши, встречается у одного ребенка на 132 000 родившихся младенцев.
Сейчас Снежана помогает больным мукополисахаридозом как президент МБОО «Хантер-синдром»: рассказывает, как получить лекарство, через благотворительные фонды находит памперсы, инвалидные коляски и ортопедическую обувь.
Право выбора
Большинство редких болезней хронические. То есть неизлечимые. Почти все приводят к инвалидизации и смерти. Орфанные препараты, если терапия от конкретного недуга все-таки существует, как правило, не излечивают болезнь полностью, а лишь снимают тяжесть симптомов и приниматься должны на протяжении всей жизни.
«С первой частью я согласилась, а вот кто такой Хантер, мне тогда было неизвестно»
Лечение двадцати четырех самых тяжелых и дорогих орфанных заболеваний финансируется государством. Если диагноз входит в знаменитый список «12 нозологий», то лекарство закупают за счет федерального бюджета, если в «Перечень 24» — за препараты платят регионы. В остальных случаях пациенты получают терапию как жизненно необходимую или по инвалидности.
Известно, что крайне важно начать лечение от мукополисахаридоза как можно раньше. Павлик получил первые свои медикаменты только в восемь. Сейчас ему девятнадцать, с восемнадцати он не говорит, хотя понимает человеческую речь.
«Конечно, мне часто пишут мамы, — признается Снежана: „Мы видели вашего ребенка в интернете. Он не разговаривает и ходит в памперсах. Зачем нужна такая жизнь?“. Но у меня короткий ответ. Что вы выберете: двенадцать лет ходить на кладбище или двенадцать лет целовать сына?».
За двенадцать лет терапии Павел не пропустил ни одного приема Элапразы. Хоть препарат и стоит около миллиона (!) рублей в неделю. Благо в Москве проблем с лечением нет.
Вопрос денег
У сына Натальи Буртаевой мукополисахаридоз IV типа, заболевание схожее, но ни в списке «12 нозологий», ни в «Перечне 24» его нет.
Даниилу уже шестнадцать. Его рост 97 сантиметров. Ходит с трудом: ноги и руки деформированы, внутренние органы тоже деформированы. С пятого класса Даня учится на дому. Нервная система при этом диагнозе не страдает, дети с ним отлично осваивают школьную программу. Но поврежденной оказывается опорно-двигательная система, страдает слух.
Препараты специальной ферментной заместительной терапии, положенной в таких случаях, существуют, но первое время были не зарегистрированы в России. Единственное лекарство, одобренное американским надзорным фармакологическим ведомством (FDA) в 2014 году, называется элосульфаза альфа — торговое название Вимизим (Vimizim).
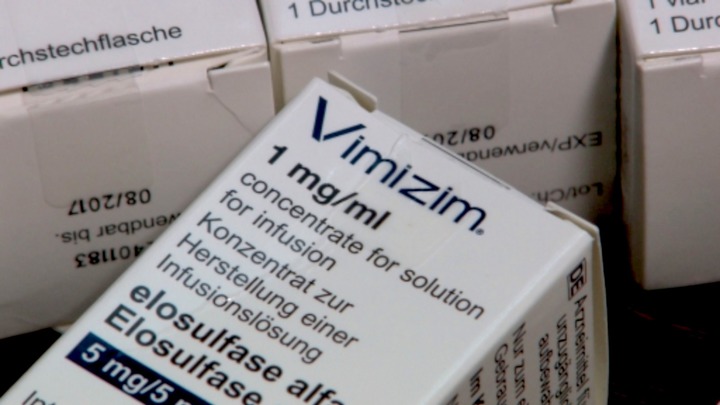
Без Вимизима больные мукополисахаридозом IV типа умирают к двадцати годам. Когда новости о его разработке только появились в специализированных СМИ, Наталья было обрадовалась. Но стоимость его годового курса превышает 60 миллионов рублей.
Минздрав Ульяновской области ожидаемо отказался приобретать препарат. Наталья подала в суд и выиграла: первый в 2016 году, второй, когда министерство подало апелляцию, — в 2017. Ни письма в Росздравнадзор и Путину, ни сюжет на «Первом канале» не помогли: Даниил до сих пор не получает лечения.
«Мы не прячемся, — твердо сообщает Наталья корреспонденту. — Даня часто гуляет на коляске. Мы с ним ездили на море, а сегодня вообще особенный день: последний звонок. Он выйдет на сцену и получит аттестат зрелости.
Добавляет: «Он у меня обычно говорит, когда я переживаю, что люди его не примут: „Мам, ты, это, не переживай. Я привык, что на меня так смотрят“».
Более того, после школы Даниил планирует учиться дальше, признается мать, вздыхая: «Не знаю, сколько он будет жить». Врачи точно ответить на этот вопрос тоже не в силах, но очевидно, что без терапии Даня проживет недолго.
«Сделано в России»
Почему орфанные препараты стоят так дорого? Фармакологические компании разрабатывают их ради прибыли. Исследования нового лекарства стоят немало, а весьма небольшой тираж препарата призван не только возместить потраченные на него миллионы, но и «вернуться» с процентами.
Если в производстве инсулина в России заинтересованы миллионы, то «целевая аудитория» орфанных препаратов даже на такую большую страну, как наша, нередко не превышает пары десятков пациентов.
Во многих странах предусмотрена государственная поддержка исследователей, разработчиков и производителей орфанных препаратов: льготы, гранты и так далее. Но в России таких программ нет.
«Мам, ты, это, не переживай. Я привык, что на меня так смотрят»
Для того чтобы снизить издержки, власти предпочитают не субсидировать или договариваться с производителем, а искать оригинальным препаратам дешевые аналоги, дженерики. К сожалению, как убеждены некоторые эксперты, они нередко отличаются по эффективности.
Насте Катасоновой двадцать один, и она не знает, сколько раз лежала в больнице: до восьми лет — раз в год, до четырнадцати — по два раза, потом — по три, сейчас — госпитализироваться приходится чуть ли не каждый месяц.
Говорит, что в больнице ей спокойно: за твоим состоянием следят специалисты. Впрочем, и тут есть поводы для волнений. У Насти муковисцидоз, редкая болезнь легких, и недавно оригинальные антибиотики заменили отечественным дженериком.
Внешне люди с редкими заболеваниями часто не отличаются от здоровых. Однажды в школе кто-то из родителей одноклассников сказал, что девочке родственники купили инвалидность, дабы та не ходила на уроки. Одноклассники в ответ на слух объявили несчастной бойкот. Теперь она о муковисцидозе говорит открыто, чтобы не возникало кривотолков.
В ноябре Насте двадцать два. Между больницами и процедурами она профессионально фотографирует, ведет личный блог, помогает с соцсетями благотворительным организациям, занимается веганским магазином.
От побочных эффектов нового дженерика — у оригинального препарата их не было — состояние Анастасии ухудшилось. «Но не пить их нельзя, иначе — захлебнешься в мокроте», — объясняет пациентка.
«Каждое утро для меня — испытание, — рассказывает она. — Мокрота за ночь отлеживается в легких. Встаю. Но не так красиво, как в фильмах, а с кашлем. За день получается стакан густой зеленой жижи. Таблетки, ингаляция, если надо на работу, приходится просыпаться за два часа до выхода». Днем опять ингаляции. Перед сном питание: через гастростому. Это специальную трубка, которая устанавливается в отверстие на животе и ведет прямо в желудок.
Екатерина Захарова, руководитель лаборатории наследственных болезней обмена веществ, председатель экспертного совета по редким болезням Всероссийского общества орфанных заболеваний, считает, что нужен активный диалог с разработчиками и производителями орфанных препаратов: «Необходимо выяснять, какие лекарства фармкомпании могут предложить бесплатно, какую минимальную стоимость могут установить. Кроме того, важно понять, какие препараты Россия может производить самостоятельно, чтобы обеспечить своих граждан: возможно, в рамках госзадания будет дешевле создать российское лекарство для небольшого числа пациентов».
Преступная забывчивость
Впрочем, не все орфанные препараты стоят миллионы. Вот только получить даже относительно недорогое лекарство нуждающимся в нем порой оказывается совсем не просто.
У Виктории Рыжковой редкое генетическое заболевание. Синдром Вильсона-Коновалова, или гепатоцеребральная дистрофия.
Медь при этой болезни не выводится из организма, а проблемы с нарушением ее обмена ведут к накоплению элемента в нервной системе, почечной, печеночной тканях и роговице, что оборачивается токсическим повреждением указанных органов.
Болезнь Гоше: орфанное заболевание в практике педиатра

Рассмотрены подходы к диагностике и лечению болезни Гоше у детей. Приведен клинический пример.
Approaches to diagnostics and treatment of Gaucher’s disease in children were considered. A clinical case is given.
В последние годы особое внимание в нашей стране уделяется проблеме ранней диагностики и лечения орфанных заболеваний.
Редкие (орфанные) заболевания — это встречающиеся с определенной частотой жизнеугрожающие или хронические прогрессирующие заболевания, приводящие без лечения к смерти или пожизненной инвалидизации пациентов. На сегодня в мире их насчитывается более 7,5 тысяч. Большинство из них обусловлено генетическими отклонениями, симптомы многих из них могут быть очевидны уже с рождения или же проявляться в детском, реже в более старшем возрасте. В 2012 г. принят Федеральный закон от 21.11.2011 № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации», в котором впервые на государственном уровне введено понятие редких (орфанных) заболеваний. В нашей стране к орфанным относятся заболевания, которые имеют распространенность не более 10 случаев заболевания на 100 000 населения (статья 44). Одним из заболеваний, включенных в группу орфанных, является болезнь Гоше (код по МКБ-10 Е75.2 — «Другие сфинголипидозы»).
Болезнь Гоше (БГ) — наиболее частая форма наследственных ферментопатий из группы лизосомных болезней накопления. Тип наследования — аутосомно-рецессивный. Присутствие двух мутантных аллелей гена ассоциируется со значительным снижением (менее 30% от нормального уровня) каталитической активности β-D-глюкозидазы (глюкоцереброзидазы), что приводит к накоплению в лизосомах макрофагов глюкоцереброзида. Ген глюкоцереброзидазы картирован на хромосоме 1q21. Частота БГ в общей популяции составляет 1:40 000–1:60 000; среди евреев-ашкенази (выходцев из Восточной Европы) — 1:450–1:800 [1]. Возраст манифестации заболевания широко варьирует — от рождения до старости.
В зависимости от клинического течения выделяют 3 типа БГ:
Тип 1 является самым частым. В отличие от 2-го и 3-го типов при данном типе нервная система в патологический процесс не вовлекается [1–4].
При БГ 1-го типа основными клиническими проявлениями являются: задержка физического и полового развития, гепатоспленомегалия, геморрагический и астенический синдромы (ассоциированы с тромбоцитопенией, анемией или панцитопенией), нарушение подвижности в суставах, патологические переломы, боли в костях (костные кризы).
Наиболее ранним признаком БГ 1-го типа заболевания является спленомегалия. Селезенка может увеличиваться в размерах в 5–80 раз. По мере прогрессирования заболевания и увеличения размеров органа возможно развитие инфарктов или разрыва селезенки с возникновением фатального кровотечения. Гепатомегалия выражена в меньшей степени и развивается, как правило, в более поздние сроки заболевания, при этом функция печени не страдает. Геморрагический синдром, обусловленный тромбоцитопенией и нарушением функции тромбоцитов, проявляется в виде развития подкожных гематом, повышенной кровоточивости слизистых оболочек, длительных кровотечений после малых оперативных вмешательств, экстракции зубов, меноррагий и пр. Поражение костно-суставной системы может протекать как в виде бессимптомной остеопении и колбообразной деформации дистальных отделов бедренных костей (колбы Эрленмейера), так и в виде тяжелейшего остеопороза, сопровождающегося множественными патологическими переломами и ишемическими некрозами, приводящим к развитию вторичных остеоартрозов и тяжелых необратимых ортопедических дефектов. У трети больных отмечаются хронические боли в костях. Они могут протекать в виде костных кризов — мучительной, интенсивной боли, чаще в области нижних или верхних конечностей, сопровождающейся гиперемией и болезненностью в области суставов, снижением двигательной активности, лихорадкой, ознобом, повышением уровня маркеров воспаления (лейкоцитоз, повышенная скорость оседания эритроцитов).
При БГ 2-го типа клинические симптомы проявляются уже на первом году жизни, часто уже в первое полугодие. Заболевание имеет быстро прогрессирующее течение и характеризуется задержкой психомоторного развития с потерей ранее приобретенных навыков, нарушением глотания, часто осложняющимся аспирационной пневмонией, тризмом, билатеральным фиксированным косоглазием, гиперрефлексией, положительным симптомом Бабинского, прогрессирующей спастичностью с ретракцией шеи, приступами тонико-клонических судорог, резистентных к традиционной противосудорожной терапии, развитием гепатоспленомегалии.
При БГ 3-го типа неврологические проявления сходны с таковыми при БГ 2-го типа, но возникают, как правило, позже (в 5–6 лет и старше) и протекают менее выраженно. Для данного типа характерны окуломоторные расстройства, экстрапирамидная и мозжечковые нарушения, генерализованные тонико-клонические судороги, миоклонии. Снижение интеллекта может проявляться от незначительных изменений вплоть до тяжелой деменции. Течение заболевания медленно прогрессирующее.
Диагностика БГ основывается на выявлении характерных клинических проявлений — задержка физического и полового развития, астенический синдром, повышенная кровоточивость, боли в костях и суставах, гепатоспленомегалия, указание в анамнезе на переломы, реже — неврологическая симптоматика в виде глазодвигательной апраксии или сходящегося косоглазия, атаксии, снижение интеллекта и др., на изменениях в клиническом анализе крови — тромбоцитопения, лейкопения, анемия или панцитопения, наличие признаков гепатоспленомегалии при УЗИ органов брюшной полости, выявление диффузного остеопороза, колбообразной деформации дистальных отделов бедренных и проксимальных отделов большеберцовых костей, очагов остеолизиса, остеосклероза и остеонекроза по данным магнитно-резонансной томографии (МРТ) или рентгенографии костей скелета, патологических переломов. Проведение морфологического исследования костного мозга позволяет выявить клетки Гоше и одновременно исключить диагноз гемобластоза или лимфопролиферативного заболевания как причины цитопении и гепатоспленомегалии.
«Золотым» стандартом диагностики БГ является определение активности β-D-глюкозидазы в лейкоцитах (сухие пятна крови) — при БГ она снижена. Также диагностическим маркером может являться повышение активности хитотриозидазы — гидролитического фермента, синтезируемого активированными макрофагами в сыворотке крови, и ДНК-диагностика.
Основой патогенетического лечения БГ является проведение ферментозаместительной терапии (ФЗТ) (показана при БГ 1-го и 3-го типов). При БГ 2-го типа она не эффективна (препарат не проникает через гематоэнцефалический барьер).
На сегодняшний день на российском фармацевтическом рынке представлены следующие препараты для ФЗТ:
В связи с гетерогенностью заболевания доза ферментного препарата для каждого больного подбирается индивидуально (от 30–60 ЕД/кг на введение при 1-м типе до 120 ЕД/кг при 3-м типе) и вводится один раз в 2 недели в/в капельно. Доза может повышаться или снижаться в зависимости от степени достижения терапевтического эффекта на основании оценки клинических проявлений.
Важно помнить, что ошибочно и необоснованно при БГ проведение спленэктомии, так как это может привести к развитию тяжелых последствий: цирроза печени, деформаций костей и суставов, костных кризов, фиброза легких, проведению повторных пункций костного мозга и других инвазивных диагностических мероприятий (биопсия печени, селезенки), оперативному лечению костных кризов (часто ошибочно рассматриваются как проявления остеомиелита), назначению кортикостероидов с целью купирования цитопенического синдрома и препаратов железа (анемия при БГ носит характер «анемии воспаления»).
В качестве клинического примера приводится история развития ребенка А., 2004 г. рождения.
Диагноз: болезнь Гоше 3-го типа (E75.2) (гомозиготная мутация L444Р в гене GBA).
Осложнения: смешанный тетрапарез. Симптоматическая фокальная эпилепсия. Косоглазие содружественное сходящееся альтернирующее. Нарушение психоречевого развития. Килевидная деформация грудной клетки. Грудной кифоз III ст. S-образный левосторонний сколиоз II ст. Остеопороз. Гепатоспленомегалия. Вторичная дилатационная кардиомиопатия. НК 0 ст.
Анамнез жизни: мальчик от первой беременности, протекавшей без осложнений, роды срочные, вес при рождении 3330 г, длина 54 см. Раннее развитие без особенностей. В возрасте 2 мес выявлено снижение уровня гемоглобина до 98 г/л, по данным УЗИ — спленомегалия. Ребенок получил два курса терапии препаратами железа, однако значимого эффекта получено не было. С одного года на фоне гипертермии и фебрильных судорог стало отмечаться сходящееся косоглазие.
В возрасте 1 г 3 мес мальчик госпитализирован в гематологическое отделение, где проведена стернальная пункция. В пунктате выявлены клетки Гоше. По данным УЗИ: гепатоспленомегалия (печень +4,5 см, селезенка до +20 см из-под края реберной дуги), в анализе крови снижение уровня гемоглобина до 90 г/л; тромбоцитов — до 114 тыс. Ед/мкл. Образцы крови направлены на генетическое исследование, в результате которого установлено снижение активности фермента β-D-глюкозидазы до 1,8 нМ/мг/час и повышение уровня хитотриозидазы до 951,6 нM/мл/час. Проведена ДНК-диагностика — обнаружена гомозиготная мутация L444Р в гене GBA. Подтвержден диагноз: болезнь Гоше.
В возрасте 1 г 6 мес (04.2006 г.) ребенок начал получать ФЗТ препаратом имиглюцеразы в дозе 40 ед/кг в/в 1 раз в 14 дней. При контрольном обследовании через 6 мес от начала терапии отмечается сокращение размеров печени (до +2,5 см) и селезенки (до +12 см из-под края реберной дуги); нормализация показателей крови (гемоглобин — 121 г/л, тромбоциты — 214 тыс. Ед/мкл). Однако с 5 лет у ребенка стали отмечаться приступы повышенной сонливости с нарушением дыхания, цианозом носогубного треугольника, судорожными движениями рук, гиперсаливацией, повторяющиеся до 5–7 раз в сутки; периодичность приступов составляла 1 раз в 7–10 дней. При обследовании по данным электроэнцефалографии патологии не выявлено, на МРТ головного мозга установлена умеренно выраженная вторичная вентрикуломегалия боковых желудочков. Ребенок консультирован психоневрологом, в терапию добавлены препараты вальпроевой кислоты пролонгированного действия, леветирацетама дигидрохлорида и топирамата. Доза препарата ФЗТ повышена до 120 ед/кг.
За 2 года комплексной терапии, включающей высокие дозы имиглюцеразы, у ребенка улучшились показатели физического развития (рис.), уменьшились размеры печени и селезенки, улучшилась структура их паренхимы, снизился уровень хитотриозидазы (до 527,5 нM/мл/час). Отмечается положительная динамика и со стороны неврологического статуса: исчезновение симптома Грефе, нарастание мышечного тонуса, улучшение координаторных и двигательных навыков, активная наработка словарного запаса (более 20 слов). Однако сохраняется сходящееся косоглазие.
.jpg)
Литература
Т. А. Бокова, доктор медицинских наук, профессор
ГБУЗ МО МОНИКИ им. М. Ф. Владимирского, Москва
Болезнь Гоше: орфанное заболевание в практике педиатра/ Т. А. Бокова
Для цитирования: Лечащий врач № 9/2019; Номера страниц в выпуске: 21-23
Теги: ранняя диагностика, ферментопатия, глюкоцереброзидаза



