УЗ «Могилевская городская больница скорой медицинской помощи»
Тема 2. Действие инсулина
Для существования организму нужна энергия.
Основным источником энергии для клеток организма является глюкоза. Глюкоза образуется при всасывании углеводов, которые человек получает с пищей.
Когда человек не ест, нормальный уровень глюкозы крови поддерживается за счет использования запасов углеводов, которые есть в организме (гликоген печени) и синтеза глюкозы из белков. Часто спрашивают, если человек не ест, почему у него повышается глюкоза крови, откуда она берется. Ответ: из гликогена печени и распада белков. Однако запасов гликогена мало (примерно 90 грамм), а синтезировать глюкозу из белка организму крайне невыгодно, поэтому при голодании организм начинает «экономить» глюкозу и отключает ее поступление в часть органов. Т.е. при голодании глюкоза поступает только в критически важные органы (мозг, сосуды, почки, нервы).
Не пропускает глюкозу в ткани при голодании инсулин. Образно говоря, на клетках некоторых органов «висит замок», который открывается инсулином. Когда инсулин открывает замок, глюкоза поступает в клетку. Эти ткани являются инсулинозависимыми, глюкоза может попасть в них только тогда, когда инсулин «даст разрешение». Инсулинозависимыми тканями являются мышцы, жировая ткань.
Но в некоторые органы глюкоза попадает без инсулина, там нет замков, дверь для глюкозы всегда открыта. Эти органы называются инсулинНЕзависимыми. Смысл действия инсулина: есть еда, можем прокормить всех, инсулин открывает двери в клетки для глюкозы. Нет еды, значит, будем кормить только самые важные органы, инсулин закрывает двери для глюкозы в менее важных органах (можно оставить «голодными» мышцы и жир, но мозг нельзя).
Но что происходит, когда нет инсулина или он дефектный? Тогда глюкоза из углеводов пищи попадает в кровь, но не может поступить в ткани. Даже если уровень глюкозы в крови высокий, инсулинозависимые ткани голодают, дверь для глюкозы в клетке закрыта на замок, ключа нет или он сломан («голод среди изобилия»).
И в то же время в инсулинНЕзависимые ткани глюкоза поступает в излишних количествах. А что излишне, то нездорово. Глюкоза начинает связываться с белками этих тканей и повреждать их. Именно из-за этого при диабете и происходит повреждение органов-мишеней (нервов, сосудов, почек и др.).
Как происходит секреция инсулина в норме?
Минимальное количество инсулина в организме вырабатывается всегда (это называется базальная секреция инсулина). Когда человек поел, всасываются углеводы, в кровь поступает глюкоза, происходит выброс инсулина (это называется пищевой или прандиальный пик), двери открываются, глюкоза идет в клетку.
В норме секреция инсулина в ответ на прием пищи происходит в 2 фазы: первую быструю фазу (1-3 минуты) и вторую медленную (до 25-30 минут).
При сахарном диабете сначала нарушается первая (быстрая) фаза.
Гормон инсулин: действие, норма содержания в крови, инсулинотерапия при сахарном диабете
Инсулин – гормон, вырабатываемый в поджелудочной железе, названной так потому, что она расположена за желудком. Инсулин позволяет нашему организму использовать глюкозу для получения энергии. Глюкоза – это разновидность сахара, моносахарид (простой углевод), который содержится в многих продуктах.
После еды или перекуса в пищеварительном тракте ферменты расщепляют сложные углеводы и превращает их в глюкозу. Затем глюкоза всасывается в кровь через слизистую оболочку тонкой кишки.
Что делает инсулин с глюкозой?
Как только глюкоза попадает в кровоток, начинает работать инсулин. Гормон заставляет клетки нашего тела поглощать сахар и использовать его для производства энергии.
Также инсулин помогает сбалансировать уровень глюкозы в крови. Когда в крови слишком много глюкозы, инсулин сигнализирует организму о том, что нужно сохранить ее избыток в печени. Накопленная в клетках печени глюкоза не высвобождается до тех пор, пока ее уровень в крови не снизится, например, между приемами пищи или когда наше тело испытывает стресс, или нуждается в дополнительной энергии (во время тренировок, напряженной мыслительной работы).
Кроме печени, по «команде» инсулина избыток глюкозы накапливается в жировой (липидной) ткани, также создавая там своеобразные «депо» для быстрой компенсации потраченной энергии.
Второстепенное действие инсулина
Гормон «работает» не только с глюкозой, но и с белками, жирами и микроэлементами:
Немного истории
Выделенный из поджелудочной железы гормон инсулин был впервые успешно применен с лечебными целями канадским врачом, физиологом, Ф.Г. Бантингом. За это в 1923 году, в возрасте 32 лет, он был удостоен Нобелевской премии по физиологии и медицине вместе со своим шотландским коллегой, профессором Маклеодом, в лаборатории которого молодой ученый проводил опыты на собаках. Открытие инсулинотерапии полностью перевернуло подходы к лечению диабета. В честь признания заслуг Бантинга, трагически погибшего в авиакатастрофе во время второй мировой войны, в день его рождения, 14 ноября, отмечают Всемирный день борьбы с диабетом.

Фредерик Грант Бантинг – ученый, впервые получивший экстракт инсулина и успешно использовавший его в лечении диабета. Один из самых молодых лауреатов Нобелевской премии за все время ее присуждения.
Инсулин: норма содержания гормона в крови, способы повышения и снижения
Очень важно, чтобы уровень инсулина в крови всегда находился в пределах нормы. Как пониженный, так и повышенный инсулин – свидетельство нарушения метаболизма (прежде всего, углеводного обмена).
Однако определение количества выделенного в кровь гормона само по себе редко бывает достаточно информативным. Дело в том, что уровень инсулина в крови в норме может колебаться в широких пределах – от 3 до 20 мкЕд/мл у детей и до 25 мкЕд/мл у взрослых. У беременных и женщин после 60-ти лет эти границы смещены, количество гормона в норме у них колеблется от 6 до 27/36 мкЕд/мл.
Поэтому определять уровень инсулина в крови нужно вместе с уровнем глюкозы (сахара) – натощак и через определенное время после приема пищи. На основании этих показателей рассчитываются определенные индексы, которые позволяют врачу обнаружить проблему и принять меры для ее коррекции либо устранения.
Повышенный инсулин натощак при нормальном сахаре может быть следствием онкологического заболевания (аденокарциномы инсулин-продуцирующих клеток поджелудочной железы).
Излишне большое количество инсулина и нормальное количество сахара через 2 часа после приема пищи выявляют у людей с так называемым предиабетом. Обменные процессы у них идут вяло, и организм пытается ускорить метаболизм глюкозы за счет выработки повышенного количества гормона. Такая интенсивная работа со временем приводит к «износу» поджелудочной железы, в результате чего она уже не может производить не только избыточное, но и достаточное количество гормона.
Как понизить инсулин в крови
Отрегулировать норму инсулина в крови при его повышении на фоне приема пищи позволяют:
Кстати, при диагностике предиабета восстановление нормального ночного сна, как правило, становится важнейшим условием для похудения, возможности переносить физические нагрузки, а, главное – для восстановления гормонального обмена и устранения инсулинрезистентности. Это позволяет затормозить переход предиабета в диабет, а в ряде случаев и полностью остановить этот процесс.
Пониженный инсулин при высоком сахаре определяется тогда, когда поджелудочная железа неспособна выработать нужное количество инсулина самостоятельно, т.е. у больного развивается сахарный диабет. Такая картина может наблюдаться как при диабете 1 типа (СД1), так и при инсулиннезависимой форме заболевания. И хотя диабет второго типа (СД2) называется инсулиннезависимым, при снижении функции поджелудочной железы ниже определенных значений таким больным, как и людям с сахарным диабетом 1 типа, показано лечение инсулином.
Инсулин как средство от диабета
Инъекции инсулина могут помочь в лечении обоих типов диабета. Инъекционный инсулин при сахарном диабете действует как замена или дополнение к инсулину вашего организма. При диабете 1-го типа необходимо вводить инсулин для контроля уровня глюкозы в крови.
Многие люди с диабетом 2-го типа могут контролировать уровень глюкозы в крови с помощью приема пероральных препаратов и/или изменения образа жизни. Однако если эти методы лечения не помогают, то, как уже говорилось выше, им тоже может понадобиться инсулин.
Терапия инсулином при диабете 1-го типа
Поскольку в организме больных с данной формой заболевания практически нет собственного инсулина, его запас необходимо пополнять каждый день для поддержания стабильного уровня сахара (глюкозы) в крови. Поэтому основное лечение больных с СД1 заключается в инсулинотерапии. Все виды инсулина дают одинаковый эффект. Они имитируют естественное повышение и понижение уровня инсулина в организме в течение дня. Состав различных типов инсулина влияет на то, как быстро и как долго они работают.
При базально-болюсном режиме терапии вводятся два вида инсулина:
Т.е., базальный инсулин поддерживает уровень сахара на желаемом уровне вне приёмов пищи и в ночные часы, чтобы обеспечить накопление фонового инсулина, а болюсный – перед приемом пищи, чтобы предотвратить скачки уровня сахара в крови после приема пищи.
Введение и дозировка
Инсулин не принимается перорально. Его необходимо водить с помощью шприца, шприц-ручки для инсулина или инсулиновой помпы. Тип инъекции и вид инсулина подбирается лечащим врачом с учетом ваших потребностей и личных предпочтений.
Поговорим про инсулин
Инсулин — это гормон поджелудочной железы, который главным образом воздействует на обмен веществ, причем в основном — на концентрацию глюкозы в крови. В своих тканях-мишенях он влияет как на мембранные, так и на внутриклеточные процессы. Некоторые из его эффектов перечислены в ниже.
Эффекты инсулина

Мембранные эффекты
Внутриклеточные эффекты
Механизм действия инсулина и влияние его на обмен

Инсулин и глюкоза
Попав в клетку, глюкоза быстро превращается в глюкозо-6-фосфат, поэтому ее внутриклеточная концентрация остается крайне низкой. Уровень глюкозы в артериальной крови в норме поддерживается в пределах 4-8 ммоль/л (72-144 мг/100 мл), так что по обе стороны клеточной мембраны всегда существует градиент ее концентраций. Несмотря на это, однако, простая диффузия обеспечивает поступление в большинство клеток лишь небольшого количества глюкозы, которого явно недостаточно для удовлетворения их метаболических потребностей (даже при возрастании концентрационного градиента, как это имеет место при высокой гипергликемии). В присутствии же инсулина проникновение декстрозы в клетки резко усиливается. Это действие инсулина проявляется лишь при наличии концентрационного градиента глюкозы, конкурентно ингибируется другими моносахаридами (например, галактозой) и следует кинетике насыщаемого процесса. Таким образом, гормон стимулирует процесс облегченной диффузии декстрозы, который осуществляется при участии чувствительных к гормону белковых транспортеров глюкозы (GLUT), расположенных на клеточной мембране. Эти транспортеры способны переносить глюкозу через клеточную мембрану в обоих направлениях, но ее поток зависит от концентрационного градиента, который направлен из внеклеточного пространства во внутриклеточное. В разных клетках найдены многочисленные GLUT, но инсулинозависимым является только один из этих белков — GLUT4, и именно он присутствует в мембранах клеток скелетных и сердечных мышц, а также жировой ткани.
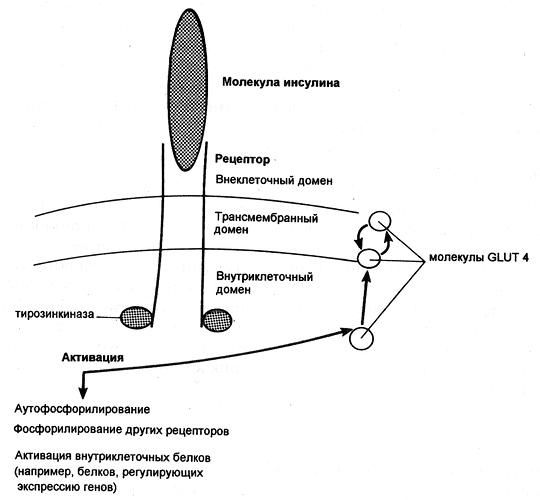
Димерный рецептор инсулина и последствия инсулиновой активации тирозинкиназы (GLUT — транспортер глюкозы)
Некоторые ткани полностью удовлетворяют свои потребности в глюкозе за счет инсулиннезависимых механизмов. Например, в клетки печени и центральной нервной системы декстроза попадает с помощью инсулиннезависимых GLUT, и поглощение этими тканями зависит только от ее уровня в крови. Кроме того, мембрану эритроцитов, клеток почек и кишечника глюкоза пересекает вместе с ионами натрия, которые поступают в клетки путем пассивной диффузии по градиенту концентрации.
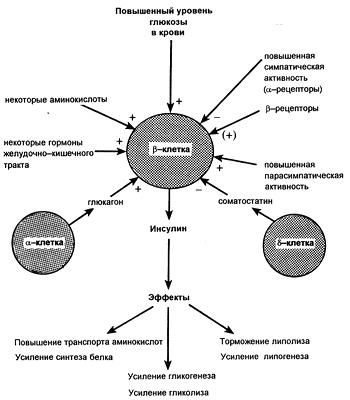
Регуляция продукции инсулина
Инсулин влияет и на внутриклеточные процессы обмена веществ. В печеночных и других клетках он стимулирует синтез гликогена, повышая активность гликогенсинтазы, что ускоряет включение гликозильных остатков в гликоген. Гормон поджелудочной железы повышает также активность печеночной глюкокиназы; этот фермент катализирует фосфорилирование глюкозы (с образованием глюкозо-6-фосфата). Одновременно гормон ингибирует печеночную фосфатазу, которая дефосфорилирует глюкозо-6-фосфат, с образованием свободной глюкозы. Такие изменения активности печеночных ферментов обусловливают снижение продукции декстрозы и наряду со стимуляцией поглощения ее периферическими клетками определяют гипогликемию, возникающее под влиянием инсулина. Возрастающая под действием последнего утилизация глюкозы в тканях обеспечивает сохранение запасов других внутриклеточных энергетических субстратов, таких как жиры и белки.
Белки и инсулин
Инсулин стимулирует не только активный транспорт аминокислот в периферические клетки, но и непосредственно синтез белка. Поскольку эти два эффекта могут не зависеть друг от друга, гормон влияет, очевидно, не только на клеточную мембрану, но и на внутриклеточные процессы. Стимуляции синтеза белка предшествует возрастание активности мРНК. Поскольку гормон с трудом проходит сквозь мембраны клеток, в механизме его ядерного эффекта должен принимать участие второй посредник. Синтез белка под действием инсулина усиливается и вследствие возрастания количества поступающих в клетку аминокислот. С другой стороны, возрастание утилизации глюкозы замедляет распад белка. Ускорение синтеза и замедление распада белка под влиянием гормона приводят к увеличению белковых запасов в интрацеллюлярном секторе.
Все эти эффекты определяют важнейшую роль инсулина в регуляции процессов роста и развития.
Инсулин и жир
Инсулин стимулирует поглощение и окисление глюкозы клетками жировой ткани. Он также стимулирует синтез липопротеиновой липазы в эндотелиальных клетках. Этот фермент катализирует гидролиз триглицеридов, связанных с липопротеинами крови, и способствует поступлению жирных кислот в адипоциты. Наряду с прямой стимуляцией липогенеза в печени и жировой ткани это приводит к увеличению запасов жира. Кроме того, инсулин ингибирует опосредуемый цАМФ липолиз, тормозя гормончувствительную внутриклеточную липопротеиновую липазу.
Инсулин и калий
Присутствие инсулина необходимо для поддержания внутриклеточной концентрации ионов калия; этот эффект, по всей вероятности, является следствием прямого влияния гормона на клеточную мембрану.
Инсулин
Инсулином называется гормон, производимый бета-клетками островков Лангерганса поджелудочной железы. Название инсулина происходит от латинского insula – остров. Эффекты инсулина
Несмотря на то, что инсулин вызывает множество эффектов в различных тканях человеческого тела, его основным эффектом является стимулирование перехода глюкозы из крови внутрь клеток, что приводит к снижению концентрации глюкозы в крови.
Другими эффектами инсулина являются стимулирование синтеза в печены и мышцах гликогена из глюкозы, увеличение создания жиров и белков, подавление активности ферментов, разрушающих жиры и белки. Таким образом, инсулин обладает анаболическим действием, поскольку усиливает образование жиров и белков, одновременно замедляя их распад.
Основной эффект инсулина заключается в усилении переноса глюкозы через клеточную мембрану внутрь клетки. Других гормонов, снижающих уровень глюкозы крови, в организме человека не существует. Основные эффекты инсулина проявляются в мышцах и жировой ткани, поэтому эти ткани называют инсулинозависимыми. Уровень глюкозы крови снижается при воздействии инсулина и повышается при воздействии т.н. гипергликемических гормонов (глюкагона, соматотропного гормона, глюкокортикоидов).
Дополнительными эффектами инсулина являются увеличение интенсивности образования гликогена, уменьшение образования глюкозы в печени, усиление поглощения клетками аминокислот, необходимых для синтеза белка. Одновременно инсулин уменьшает разрушение белков и жиров. Таким образом, общий эффект инсулина является анаболическим – направленным на формирование жировой и мышечной ткани.
Строение инсулина
Инсулин является полипептидным гормоном, состоящим из двух аминокислотных цепей: А- и В-цепи. Полипептидные цепи соединяются дисульфидными мостиками. Человеческий инсулин по структуре близок к свиному и бычьему, хотя и отличается от них одним и тремя аминокислотными остатками соответственно.
Открытие инсулина
Островки поджелудочной железы были открыты в 1869 году Паулем Лангергансом при микроскопическом исследовании структуры поджелудочной железы. В 1889 году Оскар Малиновски в Германии при удалении поджелудочной железы у собаки вызвал у нее симптомы сахарного диабета. В 1921 году Ф. Бантинг и Ч. Бест выделили из клеток островков поджелудочной железы инсулин, а Д. Коллип разработал методику его очистки.
В 1922 году инсулин впервые был введен пациенту, страдающему сахарным диабетом. Его лечебное действие показало, что такой вид терапии является наиболее эффективным. В последующие годы основные усилия ученых были направлены на организацию производства в больших количествах. В 1923 году была вручена Нобелевская премия за открытие и выделение инсулина. В последующем аминокислотная структура инсулина была полностью расшифрована Ф. Сенгером.
Синтез инсулина
В островковых клетках поджелудочной железы инсулин синтезируется в несколько этапов. На первом этапе происходит синтез молекулы предшественника инсулина – препроинсулина. На втором этапе от молекулы препроинсулина отделяется сигнальный пептид, после чего образуется проинсулин. После созревания происходит образование окончательной молекулы инсулина. На этапе созревания от молекулы проинсулина отделяется С-пептид, который не оказывает биологического действия. После отделения С-пептида формируется активная форма инсулина.
Выделение инсулина в кровь происходит при повышении уровня глюкозы в крови. Дополнительно регуляция выработки инсулина производится автономной нервной системы. Разрушение инсулина происходит в печени и почках при воздействии фермента инсулиназы.
Препараты инсулина
В настоящее время фармацевтическая промышленность производит значительное число препаратов инсулина, имеющих различные биологические эффекты. Выделяют человеческий, свиной инсулины, инсулин крупного рогатого скота. По степени очистки различают традиционные, монопиковые, монокомпонентные инсулины. По времени действия выделяют инсулины короткого и пролонгированного действия. Последние делятся на инсулины среднего, длительного и сверхдлительного срока действия. Есть также инсулины ультракороткого и депо-инсулины, выделяющиеся медленно из подкожной клетчатки.
Подбор схемы инсулинотерапии – сложное и очень ответственное мероприятие. От правильности выбора формы инсулина и схемы его дозирования зависит успешность достижения компенсации сахарного диабета и, как следствие, качество жизни пациента.
Типы сахарного диабета
В настоящее время выделяется два основных типа сахарного диабета, различающиеся по причине и механизму появления, а также по принципам лечения
Сахарный диабет 1 типа
Сахарный диабет 2 типа
Гестационный сахарный диабет при беременности
Гестационный сахарный диабет может развиваться при беременности (примерно в 4% случаев). В его основе лежит снижение способности по усвоению глюкозы
Гипогликемия
Гипогликемией называют патологическое состояние, характеризующееся снижением концентрации глюкозы в плазме крови ниже уровня 2,8 ммоль/л, протекающее с определенной клинической симптоматикой, либо менее 2,2 ммоль/л независимо от наличия или отсутствия клинических признаков
Кома при сахарном диабете
Информация о наиболее опасном осложнении сахарного диабета, требующем экстренной медицинской помощи,- коме. Описываются виды ком при сахарном диабете, их специфические признаки, тактика лечения
Синдром диабетической стопы
О диабете
Сахарный диабет — термин, объединяющий эндокринные заболеваний, характерной чертой которых является недостаточность действия гормона инсулина. Главным симптомом сахарного диабета является развитие гипергликемии – увеличения концентрации глюкозы в крови, имеющее стойкий характер
Симптомы диабета
Эффективность лечения сахарного диабета напрямую зависит от времени выявления этого заболевания. При сахарном диабете 2 типа заболевание может длительное время вызывать лишь незначительно выраженные жалобы, на которые пациент может не обращать внимания. Симптомы диабета могут быть стертыми, что затрудняет диагностику. Чем раньше поставлен правильный диагноз и начато лечение, тем меньше риск развития осложнений сахарного диабета
Анализы в СПб
Одним из важнейших этапов диагностического процесса является выполнение лабораторных анализов. Чаще всего пациентам приходится выполнять анализ крови и анализ мочи, однако нередко объектом лабораторного исследования являются и другие биологические материалы.
Консультация эндокринолога
Специалисты Северо-Западного центра эндокринологии проводят диагностику и лечение заболеваний органов эндокринной системы. Эндокринологи центра в своей работе базируются на рекомендациях Европейской ассоциации эндокринологов и Американской ассоциации клинических эндокринологов. Современные диагностические и лечебные технологии обеспечивают оптимальный результат лечения.
Анализ на гликогемоглобин
Гликогемоглобин (гликированный гемоглобин, гликозилированный гемоглобин, гемоглобин А1с) – соединение гемоглобина с глюкозой, образующееся в эритроцитах
Отзывы
Истории пациентов
Видеоотзывы: опыт обращения в Северо-Западный центр эндокринологии
Инсулин в патогенезе центральной инсулинорезистентности, сахарном диабете 2 типа и болезни Альцгеймера
Долгое время мозг считался нечувствительным к инсулину органом, однако последние исследования свидетельствуют о ключевой роли инсулиновой сигнализации в подержании гомеостаза глюкозы, контроле клеточной пролиферации и регуляции когнитивных процессов.
Авторы: Enrique Blázquez, Esther Velázquez, Verónica Hurtado-Carneiro and Juan Miguel Ruiz-Albusac
Перевод: Ирина Буянова. Редакция: Илья Левашов.
Аннотация
Инсулин может попадать в мозг как из периферических тканей, так и синтезироваться клетками центральной нервной системы (ЦНС). Молекулярные механизмы действия инсулина в ЦНС не отличаются от периферического действия гормона. Результаты последних исследований демонстрируют нейротрофический, нейромодуляторный и нейропротективный эффекты инсулина и его основополагающую роль в регуляции гомеостаза глюкозы, репродуктивной и когнитивных функций, пищевого поведения, а также пролиферации и дифференцировке клеток.
Нарушение механизмов инсулиновой сигнализации в мозге лежит в основе патогенеза центральной инсулинорезистентности, сахарного диабета 2 типа (СД2) и болезни Альцгеймера (БА). Кроме того, появляется все больше данных о связи между нарушением метаболизма глюкозы в ЦНС и риском БА.
Общими звеньями патогенеза БА и СД2 являются митохондриальная дисфункция и нарушение энергетического гомеостаза, оксидативный стресс, образование модифицированных липопротеинов низкой плотности (ЛПНП), снижение активности O-GlcNAc трансферазы, формирование амилоидных бляшек, изменение метаболизма бета-амилоидного пептида (Aβ) и повышение фосфорилирования тау-белка.
Введение
Поддержание гомеостаза глюкозы в периферических тканях – одна из основных функций инсулина. В последнее время появляется все больше данных о наличии рецепторов к инсулину (ИР) в разных отделах ЦНС и их роли в регуляции важнейших физиологических функций, включая нейрональное развитие, метаболизм глюкозы, пищевое поведение и когнитивные процессы, а также в патогенезе нейродегенеративных заболеваний (1).
Как инсулин попадает в мозг
Присутствие инсулина в нервной ткани впервые было описано в работе Havrankova et al. в 1978 году и подтверждено многочисленными исследованиями на животных (2, 3). Исследователи показали, что уровень инсулина в ЦНС не зависит от концентрации инсулина в периферических тканях и превышает его в 10-100 раз (4).
Присутствие инсулина в цереброспинальной жидкости (ЦСЖ) служит биомаркером проницаемости гематоэнцефалического барьера (ГЭБ), который определяет эффективность проникновения гормона в ЦНС и препятствует его быстрому выведению (5, 6). Считается, что основным источником инсулина в ЦНС служат специфические популяции нейронов, хотя, как отмечает Havrankova et al. (2), не стоит исключать проникновение инсулина из периферического кровотока и через ГЭБ.
Инсулин периферического происхождения
В 1967 году Margolis и Altszuler впервые высказали гипотезу о наличии механизма, опосредующего транспорт инсулина через ГЭБ (7). В эксперименте на крысах авторы работы показали, что внутривенное введение инсулина вызывает незначительное повышение его уровня в ЦСЖ. Это привело к предположению, что инсулин может проникать через ГЭБ посредством рецепторзависимого транспорта (8). Выраженное повышение уровня инсулина в крови и незначительное увеличение его концентрации в ЦСЖ в ответ на системное введение гормона указывает на наличие специальной транспортной системы, позволяющей доставлять инсулин через ГЭБ. Хотя до сих пор неизвестно, являются ли переносчики инсулина и ИР в ЦНС представителями одного семейства белков, данные многочисленных исследований демонстрируют (9) сходные физико-химические свойства (насыщаемость, специфичность, афинность, кинетику распада и иммунонейтрализацию) этих белков (10, 11). С другой стороны, активность транспортеров ГЭБ может меняться в зависимости от региональных различий в проницаемости к инсулину и уровню гормона в крови. Наиболее высокое содержание инсулина обнаружено в мосте, продолговатом мозге и гипоталамусе; самый низкий уровень гормона показан в нейронах затылочной коры и таламуса (12). Транспорт инсулина через ГЭБ регулируется различными физиологическими факторами, например, действием глюкокортикоидных гормонов (13), и обладает высокой чувствительностью к изменению энергетического гомеостаза при голодании (14), ожирении (15), гибернации (16), а также в процессе старения и у пациентов с СД и БА (17, 18).
Инсулин центрального происхождения
Биосинтез инсулина в ЦНС хорошо изучен и был продемонстрирован в ряде экспериментальных исследований.
Определение уровня С-пептида в мозге
В исследованиях post mortem показано высокое содержание иммунореактивного инсулина и С-пептида в различных структурах мозга, в несколько раз превышающее их содержание в периферической крови (19). Уровень С-пептида в сыворотке крови и структурах мозга значительно снижается после 72 часов голодания и повышается в ответ на пероральное введение глюкозы (20). Прямое доказательство связи между уровнем С-пептида и снижением количества ИР в ЦНС было получено в исследовании с участием пожилых пациентов с БА, у которых наиболее высокий уровень С-пептида был обнаружен в коре головного мозга (21).
С помощью метода гибридизации in situ удалось выделить мРНК инсулина из нейронов перивентрикулярного ядра гипоталамуса крыс (22). Кроме того, мРНК инсулина II присутствует в мозге крыс на всех этапах развития, что указывает на раннюю экспрессию гена-предшественника в мозге (23). У новорожденных кроликов показано наличие мРНК инсулина в CA1 и CA3 областях гиппокампа, зубчатой извилине и клетках гранулярного слоя обонятельной луковицы (24).
Экспериментальные подходы к работе с культурами клеток мозга
Многочисленные данные о механизмах синтеза инулина в ЦНС были получены в исследованиях на культурах клеток. Показано, что инкубация нейронов в растворе циклогексимида приводила к 80%-му снижению количества инсулин-иммунореактивных клеток в мозге крыс (25). В эмбриональных нейронах мышей было показано наличие двух форм иммунореактивного инсулина (ИРИ). Под действием трипсина про-инсулин расщепляется на компоненты, схожие по структуре с панкреатическим инсулином (26). Данные иммуногистохимического анализа и гибридизации in situ демонстрируют способность эмбриональных нейронов экспрессировать и выделять инсулин-подобные мРНК и инсулин-подобные белки, схожие по структуре с инсулином периферического происхождения (27). В мозге млекопитающих синтез иммунореактивного инсулина происходит в только нейронах ЦНС, но не в астроцитах (24).
Молекулярные механизмы, участвующие в регуляции образования и секреции инсулина в ЦНС, сходны с механизмами синтеза инсулина в периферических тканях. Одним из общих звеньев центрального и периферического путей образования инсулина является деполяризация, вызванная активацией АТФ-чувствительных K+ каналов (28). Связывание гормонов или глюкозы с рецепторами на поверхности клеточных мембран вызывает деполяризацию и стимулирует высвобождение инсулина. В культурах нейронов (но не астроцитов) циклогексимид ингибирует экзоцитоз, индуцируемый деполяризацией (29). Кроме того, в мозге взрослых крыс повышение мембранного потенциала способствует высвобождению инсулина.
Эффективность экзоцитоза инсулина во многом зависит от внутриклеточного содержания уровня кальция. Кальций-зависимый экзоцитоз специфичен для синаптических везикул нервных окончаний, что подтверждает данные о синтезе инсулина в нейронах ЦНС и его хранении в составе синаптических везикул (30).
Секреция инсулина из синаптосом повышается в ответ на увеличение уровня глюкозы, а добавление ингибитора гликолитического метаболизма – йодацетиловой кислоты (ЙК) – в культуры клеток мозга вызывает снижение высвобождения ИРИ на 50%, что подчеркивает регуляторную роль метаболизма глюкозы в секреции инсулина в ЦНС (31).
Таким образом, результаты последних исследования подтверждают наличие центрального пути синтеза инсулина (32).
Влияние инсулина на эндотелиальные клетки и ГЭБ
ГЭБ образован особым типом эндотелиальных клеток (33), которые формируют физический барьер между клетками мозга и кровотоком (17). Поверхность клеток эндотелия содержит инсулин-связывающие сайты, которые выполняют транспортную и рецепторную функции (34). Эти инсулин-связывающие сайты играют ключевую роль в регуляции внутриклеточных каскадов в клетках ГЭБ и инсулиновой сигнализации, а также способствуют повышению эффективности транспорта тирозина и триптофана (35), азидотимидина (36) и лептина (37) из периферической крови в мозг.
Кроме того, инсулин оказывает регуляторное действие на экспрессию и активность некоторых транспортных белков. Так, инсулин повышает экспрессию гликопротеина-Р (170-кДа), играющего важную роль в поддержании целостности ГЭБ (38), и подавляет экспрессию и активность белка лекарственной устойчивости рака молочной железы (39). Нейрохимические изменения в капиллярах мозга в ответ на действие инсулина реализуются через снижение активности щелочной фосфатазы (40). Вместе с тем наблюдается активация элемента антиоксидантного ответа 4 и увеличение экспрессии каталитической субъединицы глутаматцистеин лигазы (41). Инсулин-зависимое снижение активности серотониновых рецепторов подсемейства 5-HT2c в сосудистом сплетении мозга указывает на роль MAP-киназного пути в реализации эффекта серотонинового рецептора (42).
.png)
Рисунок 1. Проникновение инсулина через ГЭБ опосредовано действием белков-переносчиков
Ферменты, участвующие в деградации инсулина, также в большом количестве присутствуют в нейронах коры больших полушарий, гиппокампа, мозжечка и ствола, а также в олигодендроцитах, сосудистом сплетении и эндотелиальных клетках (43). При низком уровне пептида Aβ наблюдается увеличение активности ферментов, разрушающих инсулин, что указывает на их роль в катаболизме Aβ и может стать терапевтической мишенью при разработке подходов к лечению нейродегенеративных заболеваний (44).
Механизмы действия инсулина в ЦНС
Инсулиновые рецепторы в мозге
Ген ИР расположен на хромосоме 19p13.2–19p13.3 и содержит 22 экзона, 11 из которых кодируют α и β субъединицы белка. В результате альтернативного сплайсинга +/− экзона 11 образуются две изоформы белка-предшественника – ИР-B и ИР-A, соответственно. Этот экзон кодирует небольшую последовательность аминокислот, расположенных на С-терминальном конце внеклеточного домена α-субъединицы рецептора (45). ИР-B содержатся преимущественно в инсулин-чувствительных тканях человека – скелетных мышцах, адипоцитах и печени, в то время как рецепторы типа А преобладают в структурах ЦНС (46–48).
Гетеротетрамер ИР содержит два лиганд-связывающих сайта – внеклеточные гидрофильные α-субъединицы (15 сайтов N-гликозилирования и 37 цистеиновых остатков) и две трансмембранные β-субъединицы, связанные друг с другом дисульфидными связями. β-субъединица инсулинового рецептора обладает тирозинкиназной активностью (49).
Началу активного изучения ИР в мозге в первой половине 1970-х годов предшествовало исследование на крысах, в котором было показано снижение уровня глюкозы в крови в ответ на введение 500 μU инсулина в сонную артерию (50). Кроме того, в других исследованиях на животных было показано активное связывание меченого 125I-инсулина на мембранах клеток различных тканей (51).
Метаболизм углеводов в печени регулируется преимущественно посредством холинергической активации, нежели изменением секреторной активности островковых клеток поджелудочной железы. Впервые ИР были обнаружены в структурах мозга в 1978 году (52) и присутствовали на всех исследуемых этапах развития (53). С этих пор широкое распространение ИР в ЦНС было неоднократно подтверждено in vitro и in vivo.
Использование радиоактивной метки для оценки содержания инсулина в культурах тканей показало преобладание ИР на поверхности клеток передней части гипоталамуса и значительно более низкое количество ИР на мембранах клеток задней доли гипоталамуса, таламуса и коры больших полушарий (54). Активное связывание меченого инсулина [125I] также было показано в структурах обонятельной и лимбической систем в новой коре и корковых областях, получающих афферентные сигналы от базальных ганглиев, гиппокампе, мозжечке и сосудистом сплетении мозга, что подтверждает нейромодуляторную роль инсулина в ЦНС (55). Методами авторадиографии и компьютерной денситометрии показано преобладание ИР в зонах, связанных с обонянием, пищевым поведением и автономными функциями (56). Данные гибридизации in situ демонстрируют высокий уровень мРНК ИР в клетках гранулярного слоя обонятельной луковицы, мозжечка и зубчатой извилины, а также пирамидных клетках пириформной коры, гиппокампа, сосудистого сплетения и дугообразного ядра гипоталамуса (57).
Экспрессия мРНК ИР выше у крыс Цукера с ожирением (fa/fa) по сравнению с крысами с нормальной массой тела (Fa/−) (58). Однако связывание инсулина в культуре клеток мозга нормальных крыс не отличается от связывания гормона у крыс с стрептозоцин-индуцированным диабетом, что опровергает данные о повышении экспрессии рецепторов инсулина при СД (59).
Наряду с IR, рецепторы инсулиноподобного фактора роста (ИФР1-Р) также широко распространены в ЦНС, в частности в обонятельных и сенсорных зонах мозга и областях, отвечающих за контроль автономной регуляции, включая гипофиз (60). Кроме того, характер экспрессии ИР и ИФР1-Р в мозге крыс обладает межполушарной асимметрией и различается у самцов и самок. Предполагается, что различия в пространственном распределении этих рецепторов могут лежать в основе патогенеза некоторых психических расстройств и поведенческих различий, в частности связанных с активностью гиппокампа (например, пространственное обучение и адаптивный ответ на стресс), между мужчинами и женщинами (61).
Присутствие ИР в гипоталамусе, коре больших полушарий и мозжечке в отсутствии СД подтверждено в исследованиях post-mortem (62). Динамика связывания меченого инсулина на мембранах синаптосом коры больших полушарий меняется в процессе развития. Активное связывание инсулина с ИР в мозге обнаруживается уже на 14-й неделе внутриутробного развития, постепенно снижается к 30-й неделе и достигает минимальных значений после рождения (63).
Несмотря на различия в размере (α-субъединица ИР в мозге, ИР-А, меньше по молекулярному весу, чем α-субъединица периферического ИР, ИР-В), степени гликозилирования (выше в периферических ИР) и специфичности, кинетика и фармакологические свойства ИР в ЦНС не отличаются от свойств инсулиновых рецепторов в периферических тканях (64). С другой стороны, связывание инсулина центральными и периферическими ИР запускает разные молекулярные каскады. Так, избыток инсулина в периферических тканях вызывает снижение экспрессии ИР, и не влияет на экспрессию ИР в мозге (65). Такая гетерогенность действия центральных и периферических ИР способствует независимой и специфичной регуляции клеточных каскадов в ответ на связывание одних и тех же лигандов.
Активность экспрессии ИР также определяется типом рецептора (66). Плотность ИР на мембранах нейронов меняется на каждом этапе развития мозга. В процессе активного нейрогенеза наиболее высокий уровень ИР обнаружен в таламусе, хвостатом ядре и скорлупе, а также некоторых ядрах среднего мозга и ствола. В мозге взрослых особей плотность ИР в этих структурах значительно снижается (67).
Система инсулиновой сигнализации в мозге
Инсулиновый рецептор принадлежит семейству рецепторных тирозинкиназ. Связывание инсулина с α-субъединицей ИР в нейронах и клетках глии стимулирует фосфорилирование и активацию β-субъединицы, обладающей тирозинкиназной активностью (68). У млекопитающих механизм инсулиновой сигнализации (Рисунок 2) регулируется посредством фосфорилирования тирозиновых остатков в составе некоторых белков, включая субстраты ИР (ИРС) (69) и адапторные белки (70), которые способствуют интеграции действия рецепторных тирозинкиназ и других звеньев сигнального пути (71). Связывание инсулина вызывает интернализацию ИР в составе окаймленных везикул (72). Этот процесс играет основополагающую роль в инсулиновой сигнализации. Попадая внутрь клетки, ИР деградируют под действием внутриклеточных ферментных комплексов или снова встраиваются в мембрану.
.png)
Рисунок 2. Трансдукция сигнала и реализация физиологических эффектов инсулина и ИФР-1
Действие инсулина в ЦНС реализуется преимущественно за счет ИРС-1 и ИРС-2. ИРС-1 участвует в регуляции роста и опосредует периферическое действия инсулина, в то время как ИРС-2 играет важную роль в созревании нейронных структур, контроле массы тела, гомеостазе глюкозы и репродуктивной функции самок (74). На NH2-терминальном конце ИРС белков содержится домен гомологии плекстрина (РН), за которым следует фосфотирозин-связывающий (PTB) домен. На С-терминальном конце находятся сайты фосфорилирования тирозина и серина/треонина (75).
Сайты фосфорилирования тирозина участвуют в координации сигнальных каскадов посредством связывания SH2 доменов эффекторных (фосфоинозитид-3-киназа, PI3K; фосфатаза SHP2; тирозинкиназа Fyn) или адапторных белков (SOCS1, SOCS-3, GRB2 и др.) (70, 74). Фосфорилирование серина ИРС-1/2 с-Jun N-терминальной киназой (JNK1) и другими протеинкиназами, наоборот, подавляет фосфорилирование тирозина в ответ на связывание инсулина. Это, наряду с убиквитин-опосредованной деградацией ИРС-1/2, играет важную роль в развитии инсулинорезистентности (76, 77). С другой стороны, повышение экспрессии ИРС-2 в ответ на действие цАМФ и активации CREB способствуют усилению инсулиновой сигнализации (78).
Синапс образует физический контакт между нейронами и способствует передаче сигналов между клетками. На роль ИРС сигнализации в постнаптическом аппарате нейронов указывают данные о коэкспрессии ИР и тирозинкиназного субстрата ИР p58/53 (IRSp53) в богатых синапсами молекулярном и гранулярном слоях мозжечка, а также в синапсах изолированных клеток гиппокампа (79). Фосфорилирование IRSp53 в ответ на действе инсулина (80, 81) играет важную роль в реорганизации цитоскелета, предшествующей росту нейритов (82), а также участвует в патогенезе ряда нейродегенеративных заболеваний (83). Это подтверждается результатами исследований на животных, демонстрирующих, что нокаут гена IRSp53 вызывает нарушения когнитивных функций, выявляемых в лабиринте Морриса и тесте на распознавание новых объектов (84).
Взаимодействие ИРС и PI3K приводит к активации PI3K и последующему фосфорилированию компонента плазматической мембраны инозитола PI (4,5)P2 с образованием инозитолтрифосфата PI (3,4,5)P3. Это, в свою очередь, стимулирует ассоциацию серинтреониновой PDK киназы (3-фосфоинозитол-зависимая протеинкиназа) и протеинкиназы В (PKB или Akt) с плазматической мембраной клетки, а также активацию Akt в ответ на фосфорилирование киназами PDK1 и PDK2 (85). Этот сигнальный путь подавляется действием липидной фосфатазы PTEN или SHIP2. Фосфорилирование TSC2 (комплекс туберозного склероза 2, туберин 2) киназой Akt приводит к активации мишени рапамицина млекопитающих (mTOR) – цитоплазматической протеинкиназы, играющей ключевую роль в регуляции клеточной пролиферации и метаболизма, и таким образом опосредует связь между инсулиновой сигнализацией и чувствительностью к уровню нутриентов (70, 86). Помимо IRS/PI3K/Akt сигнального пути существует периферический путь инсулиновой сигнализации, способствующий транслокации транспортера глюкозы GLUT-4 в ответ на действие инсулина и включающий ряд молекулярных субстратов ИР – Cbl и APS. Интеграция различных белков в состав липидных рафтов индуцирует слияние везикул, содержащих GLUT-4, с плазматической мембраной клетки (71, 85).
Связывание инсулина с ИР также приводит к фосфорилированию остатков тирозина в адапторных белках Gab-1/Shp2, Shc/Grb2 и SOS/Grb2, активации G-белка Ras и запуску сигнального каскада митоген-активируемой протеинкиназы (МАР), в частности MAPK/ERK киназы (MEK) и киназы, регулируемой внеклеточными сигналами (88). МАР-киназа ERK (extracellular signal-regulated kinase) активирует ряд цитоплазматических белков, включая S6 рибосомальную киназу p90rsk (89), белки цитоскелета, фосфолипазу А2 (PLA2), а также сигнальные белки – рецепторные тирозинкиназы, рецепторы эстрогена и белки семейства SOS и STAT (сигнальный белок и активатор транскрипции). Попадая в ядро, ERK фосфорилирует факторы транскрипции – белки семейства Ets – и участвует в контроле экспрессии генов (18, 70).
Нарушение экспрессии и функциональной активности ИР в процессе развития, в том числе в результате точечных мутаций F382V (нарушение транспорта компонентов ИР к поверхности клетки), R735S (инсулинорезистентность в результате ингибирования синтеза белков-предшественников), L1018A (подавление тирозинкиназной активности рецептора) и Y960F (различные функциональные нарушения) лежит в основе патогенеза некоторых функциональных нарушений мозга (49).
Действие инсулина в ЦНС
Роль инсулина в регуляции энергетического обмена, гомеостаза глюкозы и пищевого поведения
Глюкоза – основной источник энергии для клеток мозга вне условий голодания, когда источником служат кетоновые тела (90). Наряду с метаболической, глюкоза выполняет сигнальную функцию и участвует в регуляции углеводного гомеостаза в ЦНС и периферических тканях.
Направленность действия глюкозы в ЦНС определяется присутствием двух типов рецепторов на поверхности чувствительных нейронов, играющих важную роль в регуляции пищевого поведения, энергетического обмена и углеводного гомеостаза (49). Связывание глюкозы рецепторами первого типа (glucose-excited, GE) вызывает активацию клеток, в то время как действие глюкозы на рецепторы второго типа (glucose-inhibited, GI) подавляет их активность. Сенсором глюкозы в чувствительных нейронах служит глюкокиназа, которая участвует в поддержании энергетического баланса и контроле потребления пищи (91–94).
Эффективность захвата глюкозы определяется уровнем экспрессии белков транспортеров (Таблица 1) и сенсоров глюкозы (95). Транспортный белок GLUT-1 наиболее широко представлен в ЦНС. Две изоформы белка-транспортера различаются степенью гликозилирования. В астроцитах экспрессируется изоформа с молекулярным весом 45 кДа, устойчивая и гипо- и гипергликемии; экспрессия изоформы с молекулярным весом 55 кДа встречается преимущественно в эндотелии, повышается в ответ на гипогликемию и устойчива к гипергликемии.
Функция GLUT-1 в структурах ЦНС определяется специализацией клеток, поэтому инсулин может оказывать разнонаправленное действие на активность транспортных белков (49, 96). GLUT-2, совместно с глюкокиназой и рецептором сульфонилмочевины 1 (SUR1), экспрессируется преимущественно в латеральной области гипоталамуса и некоторых его ядрах – паравентрикулярном и дугообразном (97, 98, 49, 93, 99). В нейронах мозжечка, стриатума, коры больших полушарий и гиппокампа, а также в некоторых глиальных клетках и эндотелии преобладают транспортеры глюкозы типа GLUT-3, активность которых повышается при низком уровне глюкозы.
Таблица 1. Основные изоформы транспортеров инсулина в мозге
Глиальные клетки и эндотелий сосудов
Присутствуют в большом количестве
Нейроны, глиальные клетки, танициты
Присутствуют в ограниченном количестве
Мозжечок, стриатум, кора и гиппокамп
Нейроны, глиальные клетки и клетки эндотелия
Присутствуют в большом количестве
Обонятельная луковица, зубчатая извилина гиппокампа, гипоталамус и мозжечок
Нейроны, глиальные клетки
Присутствуют в большом количестве в некоторых областях r
Глюкоза, инсулин, физическая активность
Тела нейронов и апикальные дендриты
Присутствуют в ограниченном количестве
В отличие от периферических тканей, мозг долгое время считался нечувствительным к инсулину, в том числе из-за низкого уровня транспортеров GLUT-4 в структурах ЦНС. Наиболее высокий уровень GLUT-4 обнаружен на мембране и в цитоплазматическом пуле клеток обонятельной луковицы, зубчатой извилины гиппокампа, гипоталамуса и коры больших полушарий, хотя в этих структурах он не превышает содержание GLUT-1 и GLUT-3 (102). Наиболее высокий уровень GLUT-4 обнаружен в мозжечке, где его экспрессия напрямую регулируется действием инсулина (103). В ответ на внутривенное введение глюкозы и увеличение уровня инсулина повышается встраивание цитоплазматических форм GLUT-4 в мембрану клеток мозжечка, коры и гиппокампа (104). В GE и GI нейронах гипоталамуса GLUT-4 коэкспрессируется с ИР и глюкокиназой, что способствует повышению захвата глюкозы в ответ на действие инсулина (105).
Однако инсулин-зависимая регуляция не является доминирующим механизмом контроля транспорта глюкозы. Это подтверждается данными о наличии нейронального ответа на изменение уровня глюкозы даже в отсутствии инулина (97, 98, 106). Кроме того, показано, что инсулин не влияет на захват глюкозы в гиппокампе, а связывание инсулина с ИР не вызывает увеличения AS160-зависимой транслокации GLUT-4 (104).
Транспортер GLUT-8 обнаруживается только в структурах ЦНС, в частности в возбуждающих и тормозящих нейронах гиппокампа, и при нормальных физиологических условиях и в экспериментальной модели СД1 (107) экспрессируется только на мембране тел нейронов и проксимальных участках апикальных дендритов (108, 109). Хотя физиологическая роль GLUT-8 до сих пор малоизучена, чувствительность GLUT-8 к инсулину указывает на роль этого транспортера в мобилизации субстрата в условиях дефицита глюкозы (110).
Внутри клетки GLUT-8 способствует транспорту глюкозы из шероховатого эндоплазматического ретикулума (ЭР) в цитоплазму и поддержанию гомеостаза глюкозы в клетках гиппокампа, обладающих высокой чувствительностью к гипергликемии и снижению уровня инсулина (111, 112). Введение глюкозы вызывает активацию GLUT-8 и увеличение транспорта глюкозы из цитоплазмы в шероховатый ЭР, но не стимулирует встраивание белка в мембрану.
Подавление действия инсулина в гипоталамусе или связывание инсулина с нейронами дугообразного ядра снижает регуляторное влияние инсулина на синтез глюкозы в печени (113) и может вызывать нарушение контроля глюконеогенеза (114) и гипергликемию у пациентов с СД (115, 116).
Действие инсулина на активность нейронов гипоталамуса опосредовано АТФ-чувствительными K+-каналами (117), активация которых вызывает гиперполяризацию и снижение ответа на глюкозу (118). Резекция отростков блуждающего нерва, через которые клетки печени получают афферентацию от гипоталамуса, также подавляет ингибирующее действие инсулина на образование глюкозы в гепатоцитах (116). Эти данные подтверждают результаты работы Claude Bernard (119), который в 1855 году показал, что разрушение четвертого желудочка вызывает глюкозурию у мышей.
Популяции нейронов, участвующих в поддержании энергетического гомеостаза, расположены преимущественно в гипоталамических центрах голода и насыщения. Как и GLUT-2, глюкокиназа (92–94), AMPK и PASK и рецепторы орексигенных и анорексигенных молекул, которые продуцируются клетками этих ядер, выступают в качестве сенсоров энергетического обмена, генерируя интегрированный ответ на афферентную стимуляцию при изменениях энергетического гомеостаза или дефиците ресурсов. Гипоталамические центры голода и насыщения детектируют сигналы об уровне глюкозы и передают их в другие зоны мозга, что приводит к активации пищевого поведения. Активация PI3K каскада является общим звеном сигнального каскада инсулина, лептина и серотонина, и играет важную роль в патогенезе инсулинорезистентности и контроле потребления пищи. В периферических тканях инсулин стимулирует анаболические процессы, в то время как в структурах ЦНС он проявляет анорексигенные свойства и выполняет катаболическую роль (120). Кроме того, в гипоталамусе инсулин активирует JAK2 и SHT3 и усиливает действие лептина (121). Механизмы резистентности нейронов гипоталамуса к инсулину и лептину у мышей с СД2 открывают новые возможности в изучении механизмов инсулинорезистентности при СД2 (33).
Роль инсулина в регуляции полового поведения
Репродуктивная способность во многом зависит от энергетического метаболизма, и нарушение гомеостаза глюкозы может нарушать нормальную регуляцию полового поведения (122), изменяя активность гипоталамо-гипофизарно-гонадальной оси (123). Достаточный уровень энергетических ресурсов способствует нормальному функционированию репродуктивной системы и повышает выживаемость потомства.
Воздействие низких концентраций инсулина на клетки гипоталамуса стимулирует секрецию рилизинг-фактора лютеинизирующего гормона (ЛГ-РГ) и зависит от гомеостаза глюкозы (124), что подтверждает его роль в регуляции репродуктивной функции. С другой стороны, воздействие инсулина в высокой концентрации не влияет на секрецию ЛГ-РГ (124). Введение инсулина в культуру клеток коры больших полушарий также повышает частоту импульсной секреции лютеинизирующего гормона (ЛГ), в то время как при введении глюкозы такой эффект не наблюдается (125). У мышей с СД низкий уровень инсулина в ЦНС вызывает снижение высвобождения ЛГ (124), которое восстанавливается при введении инсулина в мозг или периферические ткани (126). В других исследованиях показано, что низкий уровень инсулина у крыс с СД способствует снижению секреции гонадотропин-рилизинг гормона (ГнРГ) в гипоталамусе и подавлению ответа гонадотропных клеток гипофиза на действие ГнРГ (126). Таким образом, инсулин играет важную роль в контроле импульсной секреции ГнРГ (127). Однако авторы отмечают, что секреция ЛГ зависит не только от действия инсулина, но и от содержания глюкозы. Это опосредовано детекторами глюкозы в гипоталамусе, которые также могут влиять на секрецию ГнРГ, независимо от уровня инсулина (128).
Влияние инсулина на клеточную пролиферацию и дифференцировку
Трофическая функция инсулина в ЦНС реализуется через регуляцию клеточной пролиферации, дифференцировки и роста нейритов в процессе эмбрионального развития. Внутривенное введение инсулина повышает активность орнитин декарбоксилазы в мозге новорожденных крыс, которая служит маркером нейронального развития (129). Участие инсулина в процессах пролиферации подтверждается данными о повышении количества ИР в период активной клеточной дифференцировки (130). Действие ИР на созревание (131) мозга, и также рост и регенерацию аксонов (132) реализуется посредством ИРС-2.
Нейротрофическая функция инсулина была продемонстрирована в исследованиях in vitro. В мозге крыс инсулин стимулирует пролиферацию (133), а в культуре нейронов переднего мозга курицы активирует рост и дифференцировку нейронов (134). ИР, участвующие в реализации эффекта инсулина на нейрональный рост и развитие, также найдены на мембранах глиальных клеток (135, 136), в которых направленность их действия определяется типом клеток. Кроме того, инсулин и ИФР2 стимулируют активность фактора роста нервов (ФРН), необходимого для роста нейритов (137). Действие инсулина на развитие ЦНС зависит от присутствия астроцитов (138), в которых наблюдается активная пролиферация белков сигнального каскада инсулина (139, 140).
В культуре эмбриональных клеток инсулин повышает фосфорилирование рибосомального белка S6 (136) и активность протеинкиназы-эпсилон в цитоплазме клетки (141), стимулируя рост нейритов (142, 143).
Механизм действия инсулина на нейрональное развитие также включает другие ферменты, в том числе фосфатидилинозитол-3-киназу (PI3K) (144). Активация сигнального пути PI3K/mTOR в ответ на действие инсулина повышает экспрессию белка постсинаптической плотности PSD-95 в нейронах CA1 и способствует формированию дендритных шипиков и возбуждающих синапсов в клетках гиппокампа (145, 146).
В инсулин-зависимой регуляции роста нейритов также участвует тау-белок, ассоциированный с микротрубочками. Эффект тау-белка на аксональный рост реализуется через PI3K/mTOR каскад и сопровождается повышением экспрессии мРНК и уровня тубулина (147).
Эндогенный инсулин способствует формированию нейрофиламентов (148) и участвует в регуляции пролиферации и дифференцировки плюрипотентных стволовых клеток. Недостаточность инсулина вызывает неапоптотическую клеточную гибель (130), а снижение активности PI3K/Akt каскада нарушает процесс дифференцировки стволовых клеток человека (hNSC), которые, в отличие от стволовых клеток грызунов, обладают высокой чувствительностью к инсулину и функционируют в узком диапазоне концентрации гормона (149). Таким образом, инсулин играет ключевую роль в интеграции нейрональной активности и регуляции энергетического гомеостаза, определяющих каждый этап клеточной дифференцировки (150).
Нейропротективный эффект инсулина
Нейропротективный эффект инсулина проявляется в подавлении апоптоза, инактивации β-амилоидного белка, снижении оксидативного центра и ишемии. Важную роль в реализации антиапоптотического эффекта инсулина играет активность PI3K/Akt/mTOR пути и белка p70SK. Ингибирование mTOR рампамицином снижает антиапоптотический эффект инсулина, что указывает на важную роль PI3K/Akt/mTOR пути и p70SK белка в реализации этого эффекта (151). Кроме того, инсулин снижает образование Aβ фибрилл, предотвращая клеточную гибель в результате накопления β-амилоида (152).
Образование активных форм кислорода (АФК) в результате оксидативного стресса индуцирует окисление липидов и белков, вызывая нарушения функциональной активности GLUT-3 и захвата глюкозы. Это приводит к накоплению лактата, ацидозу и митохондриальной дисфункции (153). В условиях оксидативного стресса инсулин стимулирует поглощение глюкозы и образование пирувата, способствуя восстановлению внутриклеточной концентрации АТФ (154). Кроме того, инсулин предотвращает внесинаптическое накопление глутамата и гама-аминомасляной кислоты (ГАМК) в результате снижения захвата этих нейромедиаторов при оксидативном стрессе (155) и стимулирует накопление мочевой кислоты, обладающей антиоксидантной активностью (156).
Антиишемическое действие инсулина реализуется с помощью двух основных механизмов – прямого действия на нейрональные рецепторы и повышения уровня глюкозы в периферических тканях (157). При транзиторной ишемии инсулин повышает уровень ГАМК – главного тормозного нейромедиатора в мозге – во внеклеточном пространстве (158).
Стимуляция Na+/K+ АТФазы в ответ на действие инсулина приводит к снижению внеклеточной концентрации K+ и внутриклеточной концентрации Na+, способствуя уменьшению нейрональной активности и метаболических потребностей клеток, предотвращению накопления жидкости и образованию постишемического отека тканей, а также снижению уровня лактата (159, 160).
С другой стороны, реперфузия при ишемии мозга стимулирует фосфорилирование JNK1/2, экспрессию Bcl-2 и деградацию каспазы-3 в гиппокампе крыс, что указывает на важность интегрированной активности Akt и JNK1/2 в реализации противоишемического действия инсулина.
Нейромодуляторный эффект инсулина
Нейромодуляторное действие инсулина реализуется через регуляцию активности ионных токов и контроль уровня и действия нейромедиаторов. Электрофизиологический эффект инсулина in vivo опосредован действием ГАМК, которое снижается в ответ на введение ингибиторов ИР (161). Кроме того, рецепторы ГАМК являются одним из основных субстратов Akt фосфорилирования, что подтверждает роль инсулина в регуляции плотности рецепторов ГАМК на постсинаптической мембране (162).
Изменение активности ионных каналов в ответ на действие инсулина способствует реорганизации ионных токов и изменению активности нейронов. В нейронах гипоталамуса инсулин повышает активацию K+АТФ каналов, что приводит к гиперполяризации мембраны и торможению нейрональной активности (163). Кроме того, инсулин стимулирует Na+/K+ АТФазу и вызывает увеличение внутриклеточной концентрации Ca2+, стимулируя высвобождение нейропептидов (164).
α2-адренергических рецепторов в нейронах гипоталамуса (169). Помимо этого, инсулин стимулирует захват аминокислот, необходимых для синтеза нейромедиаторов (170).
Влияние инсулина на когнитивные функции и память
Внутривенное введение, внутрицеребровентрикулярная или внутригиппокампальная инфузия инсулина связаны с улучшением когнитивных функций и памяти (171), которые сопровождаются увеличением экспрессии ИР и активацией сигнальных каскадов в гиппокампе (172). Инсулин также способствует восстановлению памяти при ишемическом поражении мозга (173).
Введение низких доз стрептозоцина в мозг вызывает центральную инсулинорезистентность подавляет активность инсулина и приводит к нарушениям памяти и поведения (174). СД1 и СД2 повышают риск когнитивных нарушений и развития БА у пожилых пациентов. В эпидемиологических исследования показано, что введение инсулина больным с СД1/СД2 и БА улучшает формирование памяти и способствует поддержанию уровня глюкозы (175). Системное введение инсулина здоровым испытуемым в условиях эугликемии и гиперинсулинемии улучшает показатели вербальной памяти и селективного внимания.
Ключевую роль в формировании кратковременной и долговременной памяти играют процессы долговременной потенциации (ДП) и долговременной депрессии (ДД) (176). ДП возникает в ответ на длительную и синхронную активацию пресинаптических и постсинаптических нейронов, способствующую поддержанию деполяризации постсинаптической мембраны в результате повышения уровня Ca2+ и консолидации. ДД является компенсаторным механизмом и вызывает снижение эффективности нейрональной передачи в ответ на интенсивное воздействие стимула. Наряду с ДП и ДД, важную роль в процессах обучения и памяти играет пластичность дендритных шипиков и реорганизация цитоскелета, опосредованные действием глутамата на AMPA и NMDA рецепторы. Активность рецепторов регулируется изменением их плотности на клеточной мембране или ковалентной модификацией белковых субъединиц. В процессе ДП наблюдается увеличение плотности AMPA рецепторов на постсинаптической мембране, в то время как ДД приводит к снижению количества рецепторов. Фосфорилирование и дефосфорилирование глутаматных рецепторов при ДП и ДД играет важную роль в контроле эффективности нейронной передачи (177).
Инсулин вызывает снижение плотности AMPA рецепторов на постсинаптической мембране, способствуя ДД. Этот процесс во многом зависит от уровня фосфорилирования ИР, активности PI3К и синтеза белка (178). Кроме того, инсулин стимулирует фосфорилирование субъединицы GluR2 AMPA рецепторов в нейронах гиппокампа, способствуя их интернализации и снижению возбудимости клеток (179).
Важную роль в реализации действия инсулина на процессы обучения и памяти играет ГАМК. Инсулин стимулирует транслокацию и экспрессию рецепторов ГАМК на постсинаптической мембране, в то время как подавление активности PI3K снижает этот эффект (180).
Стимулирующее действие ИФР1 на активность нейронов в гиппокампе крыс реализуется с участием AMPA рецепторов и PI3К (181). О роли соматотропного гормона (СГ) в ДП и формировании памяти свидетельствует повышение экспрессии NMDA рецепторов в гиппокампе (182).
Воспаление и инсулинорезистентность
У пациентов с ожирением и СД2 воспаление является ключевым звеном патогенеза инсулинорезистентности в ЦНС и периферических тканях и повышает риск развития когнитивных нарушений и БА (183). У пациентов с БА обнаружено увеличение уровня интерлейкина IL-6 в ЦСЖ (184), что указывает на роль воспаления в аккумуляции β-амилоидного пептида Aβ (185). Результаты экспериментальных исследований демонстрируют противовоспалительное действие инсулина у пациентов с БА (186).
Введение липополисахарида приводит к увеличению уровня С-реактивного белка и провоспалительных цитокинов IL-1β, IL-6 и TNF в плазме крови (187). TNFα и IL-6, в свою очередь, стимулируют активацию NFkβ и транскрипцию провоспалительных факторов TNFα, IL-6 и IL-1b (188). Это приводит к нарушению синаптической пластичности в гиппокампе и формированию пространственной памяти (189).
Инсулинорезистентность периферических тканей у пациентов с ожирением связана с увеличением уровня провоспалительных цитокинов и свободных жирных кислот. Хроническое воспаление также способствует развитию инсулинорезистености, СД2 и БА (190). В частности, повышение уровня TNFα и Aβ в мозге пациентов с гиперинсулинемией стимулирует образование амилоидных бляшек (191). Среди пациентов с ожирением и БА наблюдается снижение концентрации инсулина в мозге, что указывает на нарушение транспорта инсулина через ГЭБ и снижение чувствительности нейронов к действию гормона (192).
В зависимости от типа рецепторов – TNF-R1 или TNF-R2 – TNF-α проявляет нейротоксический или нейропротективный эффект. TNF-R1 оказывает проапоптотическое действие, в то время как TNF-R2 предотвращает клеточную гибель. У пациентов с БА, диабетом и нарушением толерантности к глюкозе наблюдается увеличение уровня TNF-R1 и снижение уровня TNF-R2 (193, 194), который нормализуется после трех недель высококалорийной диеты (195). Накопление конечных продуктов гликирования, оксидативный стресс и повреждение клеточных структур способствуют нарушению когнитивных функций у пациентов с СД (196).
Нарушение каскадов инсулиновой сигнализации, в частности PI3K/Akt и киназы-3 гликогенсинтетазы GSK-3, связано с повышением воспаления и инсулинорезистентностью (197). Известно, что PI3K подавляет образование IL-12 в дендритных клетках, а GSK-3 способствует гиперфосфорилированию и регуляции метаболизма Aβ (198).
Активация ИР приводит к увеличению фосфорилирования и подавлению активности GSK-3β (199). У пациентов с БА и СД2 наблюдается повышение активности GSK-3β и увеличение фосфорилирования ИР и ИРС-1 (200). Кроме того, сигнальный каскад PI3K/Akt/GSK-3 играет важную роль в активации STAT-3 в глиальных клетках (201): ингибирование GSK-3 вызывает повышение уровня противовоспалительных цитокинов, в частности IL-10, и снижение образования провоспалительных факторов – IL-1β, IL-6 и IFN-γ (202).
Известно, что воспаление играет ключевую роль в патогенезе БА, способствуя повышению продукции провоспалительных факторов Т-лимфоцитами, моноцитами и клетками глии (203, 204). Данные исследований на животных показывают, что подавление продукции провоспалительных цитокинов позволяет снизить выраженность симптомов инсулинорезистентности (205). Хотя воспаление не участвует в развитии острой инсулинорезистентности, которая развивается в ответ на рацион с высоким содержанием жира (206), оно оказывает значительное влияние на развитие хронической инсулинорезистентности (207).
Роль TNF-α в патогенезе инсулинорезистентности реализуется через подавление тирозинкиназной активности ИР и снижение активности SOCS-3, приводя к инактивации ИРС-1 (208). Кроме того, активация JNK в ответ на повышение уровня факторов воспаления и свободных жирных кислот повышает фосфорилирование ИРС-1 и снижает эффективность инсулиновой сигнализации, способствуя увеличению уровня воспаления и развитию инсулинорезистентности. Как показывают исследования на животных, инактивация JNK и подавление воспалительного ответа позволяют избежать негативных метаболических эффектов, вызванных гиперлипидемией (210, 211, 212).
Внутрирецебровентрикулярная инфузия TNF-α стимулирует воспаление в гипоталамусе, повышение уровня инсулина и нарушение инсулиновой сигнализации в периферических тканях. Считается, что центральная инсулинорезистентность служит адаптивным механизмом при высококалорийной диете и сопровождается нарушением гомеостаза глюкозы. Повышение уровня свободных жирных кислот служит сигналом для активации высвобождения провоспалительных цитокинов, подавляющих действие инсулина (213).
У пациентов с БА показано нарушение инсулиновой сигнализации, снижение тирозинкиназной активности ИР, подавление экспрессии мРНК инсулина и ИФР1 и их рецепторов (214). Это вызывает снижение эффективности сигнальных каскадов инсулина (IR/IRS-1/PI3K) и ИРФ1 (IGF1R/IRS-2/PI3K). Уменьшение ответа на действие инсулина опосредовано фосфорилированием остатков серина (IRS-1 pS616, IRS-1 pS636/639) и инактивацией ИРС-1. При умеренных и выраженных когнитивных нарушениях наблюдается повышение маркеров инсулинорезистентности, независимо от уровня аполипопротеина E (APOE-4) (104). Снижение чувствительности к инсулину также сопровождается повышенным накоплением Aβ, который, в свою очередь, оказывает тормозящее действие на экспрессию и функциональную активность гормона (215). Пептид Aβ проявляет антагонистическое действие по отношению к инсулину, конкурентно связываясь и снижая аутофосфорилирование ИР (216, 217). Это может препятствовать реализации нейропротективного эффекта инсулина у пациентов с БА и СД2 и повышать риск когнитивных нарушений (218). Резистентность к инсулину и ИФР1 служит ранним биомаркером БА и указывает на дисфункцию ИРС-I, вызванную действием Aβ (104).
Сахарный диабет и болезнь Альцгеймера
Результаты последних исследований указывают на ключевую роль инсулина в регуляции нейрональной активности и тесную связь между БА и СД2 (219) (Рисунок 3). БА – самое распространенное нейродегенеративное заболевание, которое сопровождается потерей памяти и прогрессирующей деменцией. В настоящее время более 30 миллионов людей страдают БА, а к 2040 году ожидается рост встречаемости заболевания до 120 миллионов (197).
Важным звеном патогенеза БА является накопление амилоидных бляшек и нейрофибриллярных кубков – агрегатов гиперфосфорилированного тау-белка, а также церебральная амилоидная ангиопатия и нейродегенерация (220).
СД2 сопровождается нарушением секреции инсулина и инсулинорезистентностью. В 2010 году в мире насчитывалось 250 миллионов пациентов с сахарным диабетом, 90% из которых страдали СД2 (221). Наряду с ожирением, старение является одним из ключевых факторов риска БА и СД2.
.png)
Рисунок 3. Нарушение инсулиновой сигнализации в патогенезе болезни Альцгеймера. Aβ, β-амилоидный пептид; GLUT-3, транспортер глюкозы 3; GSK-3β, киназа-3 β гликогенсинтетазы; NFT, нейрофибриллярные клубки; PI3K, фосфатидилинозитол-3-киназа.
Инсулинорезистентность, воспаление, накопление амилоидного пептида и когнитивные нарушения являются общими звеньями патогенеза БА и СД2. Наряду с периферической инсулинорезистентностью, в патогенезе этих заболеваний также участвует резистентность к ИФР1 и нарушение функциональной активности ИРС-1 и ИРС-2.
Инсулинорезистентность в периферических тканях предшествует манифестации СД2, что представляет собой важное терапевтическое окно для своевременного лечения (222). Особый интерес вызывает вопрос о роли периферической и центральной инсулинорезистентности в патогенезе БА и СД2. Отсутствие СД2 у пациентов с БА может быть связано с поздней манифестацией симптомов диабета, хотя проявление симптомов когнитивной дисфункции также обнаруживается на поздних стадиях болезни.
Выделают четыре стадии прогрессирования БА. На первой стадии болезнь не сопровождается выраженными симптомами; на преклинической стадии заболевания обнаруживаются патофизиологические изменения на фоне отсутствия выраженных когнитивных нарушений; третья стадия предшествует развитию деменции и сопровождается умеренными когнитивными нарушениями; последняя стадия БА характеризуется тяжелой когнитивной дисфункцией и деменцией (223, 224).
Изменение уровня инсулина и нарушение метаболизма глюкозы повышают риск развития деменции (225, 226). Результаты последних исследований демонстрируют повышение экспрессии маркеров СД2 у пациентов с БА (227) и подтверждают связь между нарушением инсулиновой сигнализации и снижением когнитивных функций. В настоящее время центральная инсулинорезистентность, сопровождающаяся когнитивными нарушениями, рассматривается как отдельная форма СД – сахарный диабет 3 типа (СД3).
Однако эта форма диабета не может считаться классическим проявлением заболевания, так как у пациентов с БА не наблюдается гипергликемии, характерной для СД1 и СД2. Кроме того, в отличие от мышечной и жировой ткани и печени, инсулин не оказывает стимулирующего влияния на захват глюкозы клетками мозга (228). Центральная инсулинорезистентность может развиваться в ответ на изменение гомеостаза глюкозы в периферических тканях и сопровождаться повышением уровня Aβ и транспорта глюкозы (230). Таким образом, данные указывают на центральную роль инсулинорезистентности в манифестации БА и СД2, которой предшествует снижение чувствительности к инсулину в периферических тканях.
Переход от преддиабета к диабету длится от 10 до 15 лет и наиболее широко представлен среди пожилых людей, которые находятся в группе повышенного риска развития БА.
Преддиабет и диабет обнаруживается более чем у 81% пациентов с БА (226). Долгий период развития диабета из преддиабета объясняет, почему инсулинорезистентность у пациентов с БА не всегда сопровождается симптомами СД.
Результаты экспериментальных и эпидемиологических исследований подтверждают связь между инсулинорезистентностью при СД и риском деменции (231). Гиперинсулинемия и гипергликемия, вызванные снижением чувствительности к инсулину, способствуют образованию нейритных бляшек (233). По данным эпидемиологических исследований, с возрастом повышается вероятность коморбидного течения БА и СД, независимо от наличия сердечно-сосудистых заболеваний (234).
В то время как влияние СД2 на активность ЦНС хорошо изучено, существует гораздо меньше данных о центральных эффектах СД1 (235). Рядом исследований показана роль СД1 в нарушении процессов памяти, обучения и когнитивной гибкости (235–242). В основе этого эффекта лежит снижение количества дендритов в сером веществе мозга у пациентов с СД1 (245). Инсулиновая терапия, наоборот, способствует улучшению когнитивных функций (243, 244) и восстановлению морфологической структуры клеток (246). У пациентов с СД1 и СД2 инсулин и ИФР1 предотвращают атрофию головного мозга и когнитивную дисфункцию (247).
Однако существуют данные, опровергающие связь между БА и СД (248, 249). При прогрессирующей форме БА и отсутствии аллеля APOE-4 наблюдается снижение уровня инсулина в ЦСЖ на фоне повышения содежания гормона в крови (197). Увеличение уровня инсулина в плазме крови служит маркером инсулинорезистентности, в то время как снижение уровня инсулина в ЦСЖ указывает на нарушение метаболизма и снижение транспорта инсулина через ГЭБ.
Общими звеньями патогенеза БА и СД являются митохондриальная дисфункция, оксидативный стресс, нарушение метаболизма глюкозы, и образование модифицированных форм ЛПНП (197). Инсулин предотвращает инактивацию окислительного фосфорилирования в митохондриях и обладает протективным эффектом в отношении Aβ и окислительного стресса, в то время как при СД наблюдается снижение активности системы антиоксидантной защиты и повышение чувствительности клеток к действию токсических олигомеров амилоидного белка (250).
Независимо от наличия диабета, нарушение метаболизма глюкозы наблюдаются у большинства пациентов с БА (223), преимущественно в височно-теменной, задней поясной и лобной областях коры. Введение глюкозы стимулирует действие инсулина и способствует улучшению показателей памяти (251, 252).
Недостаточность метаболизма глюкозы связана с нарушением мембранного транспорта, энергетического гомеостаза и метаболизма тиамина. У пациентов с БА наблюдается снижение экспрессии GLUT-1 и GLUT-3 (253), особенно в нейронах коры больших полушарий и зубчатой извилине гиппокампа. Согласно результатам работы Liu et al. (254), снижение метаболизма глюкозы происходит в результате подавления экспрессии транспортных белков и O-GlcN-ацетилирования, повышения фосфорилирования тау-белка и образования нейрофибриллярных бляшек.
БА сопровождается изменением функциональной активности ключевых ферментов цикла Кребса и пентозофосфатного пути: пируватдегидрогеназного комплекса, α- кетоглутаратдегидрогеназного комплекса, транскетолазы и нуклеозид-дифосфатазы (255). Эти данные подтверждают роль митохондриальной дисфункции и изменения метаболизма тиамина в нарушении гомеостаза глюкозы в мозге при БА.
Недостаточность метаболизма глюкозы служит предиктором патофизиологических изменений, ассоциированных с СД (222). Согласно гипотезе Chen и Zhong, нарушение гомеостаза глюкозы и метаболизма тиамина и инсулинорезистентность способствуют агрегации пептида Aβ и гиперфосфорилированию тау-белка, занимая центральное место в патогенезе БА. Эти патофизиологические каскады индуцируют высвобождение провоспалительных факторов, вызывают митохондриальную дисфункцию и оксидативный стресс, повышают уровень конечных продуктов гликирования, апоптоз, эксайтотоксичность и активность протеинкиназ (223).
Общим звеном патогенеза БА и СД также является нарушение метаболизма холестерина. Гиперхолестеринемия вызывает изменение активности островковых клеток поджелудочной железы и снижение секреции инсулина (256, 257). Повышение уровня ЛПНП способствует образованию Aβ и его встраиванию в клеточные мембраны (10). Связывание Aβ с липопротеинами катализирует образование оксихолестерина, обладающего выраженным нейротоксическим действием и способствующего подавлению ERK/Akt каскада (258, 259). Гипергликемия и гиперинсулинемия стимулируют O-GlcN-ацетилирование белков каскада инсулиновой сигнализации и способствуют развитию инсулинорезистентности (260, 261). В образцах лобных областей коры пациентов с СД2 и БА наблюдается снижение активности инсулин/PI3K/Akt сигнального пути и (262) и O-GlcNAc трансферазы, наряду с повышением GSK-3β кальпаин-1-зависимой регуляции, что способствует гиперфосфорилированию белков и нейродегенерации (262).
Образование амилоидных агрегатов, изменение метаболизма Aβ и гиперфосфорилирование тау-белка служат связующим звеном в патогенезе БА и СД. В клетках островков Лангерганса могут формироваться скопления амилоидного белка IAPP, который сходен по структуре с пептидом Aβ и обладает токсическим действием на клетки поджелудочной железы (263, 224). Снижение активности шаперонов, препятствующих агрегации IAPP и Aβ, способствует образованию амилоидных и нейритных бляшек в островковых клетках и нейронах у пациентов с БА и СД (226).
Протеазы играют важную роль в метаболизме и предотвращении накопления продуктов распада Aβ. В частности, металлопептидаза IDE способствует повышению активности инсулина, ИФР и глюкагона. Нарушение инсулиновой сигнализации сопровождается выраженным увеличением уровня фосфорилированного тау-белка и пептида Aβ и способствует клеточной гибели и нейродегенерации, особенно у пациентов с СД2 (264). Повышение уровня инсулина в ЦНС оказывает стимулирующее действие на функциональную активность IDE (265, 266), а также способствует интернализации олигомеров Aβ и предотвращает их связывание с мембраной нейронов.
Важную роль в патогенезе БА играет формирование нейрофибриллярных клубков, которые представляют собой агрегаты гиперфосфорилированного тау-белка (267).
Инсулинорезистентность и периферическая гиперинсулинемия при СД2 приводят к снижению транспорта инсулина через ГЭБ, а системная инсулиновая недостаточность при СД1 повышает фосфорилирование тау-белка (235, 268).
Таким образом, центральная инсулинорезистентность вызывает повреждение нейронов гиппокампа и коры больших полушарий и служит общим звеном патогенеза СД2 и БА. Наряду с нарушением инсулиновой сигнализации, развитию БА могут способствовать вирусные, бактериальные и грибковые инфекции (269, 270, 271). Однако во всех случаях БА сопровождается прогрессирующей нейродегенерацией.
Терапевтические подходы
Понимание механизмов регуляции когнитивных и метаболических функций открывает новые возможности в терапии инсулинорезистентности при СД2 и БА. Ни один из современных терапевтических подходов, направленных на известные звенья патогенеза нейродегенеративных заболеваний – агрегаты амилоидного пептида (272), гиперфосфорилирование тау-белка (273) и метаболизм тиамина, а также использование антиоксидантов и нейропротектиных факторов (274, 275, 276), не позволяет достигнуть высокой эффективности лечения.
В последнее время все большую популярность набирают мультитаргетные подходы, нацеленные на регуляцию молекулярных каскадов, опосредующих патогенетический эффект инсулинорезистентности (276). В частности, наиболее известными молекулярными агентами, способствующими повышению секреции инсулина, являются метформин, антагонисты рецептора, активируемого пролифераторами пероксисом PPARγ, а также миметики инкретина, глюкагоноподобный пептид-1 (ГПП-1) и гастрический ингибирующий полипептид. Наряду с этим показана высокая эффективность интраназального введения инсулина и ГПП-1 в терапии СД2 и при умеренных когнитивных нарушениях (224).
Метформин, представитель класса бигуанидов, способствует поддержанию нормального уровня инсулина натощак и контролирует образование глюкозы в печени. В последнее время появляется все больше данных о способности метформина проникать через ГЭБ, повышая чувствительность к инсулину в структурах ЦНС (277) и способствуя снижению риска деменции у пациентов с СД (278).
Использование агонистов PPARγ, в частности росиглитазона, позволяет добиться значимого улучшения чувствительности к инсулину (279), увеличения функциональной активности адипоцитов, повышения транспорта триглицеридов и жирных кислот из печени и мышц, а также предотвращения накопления Aβ и снижения уровня воспаления (280, 281). Это приводит к нормализации уровня инсулина и улучшению внимания и памяти на ранних этапах патогенеза БА (282). Несмотря на высокую эффективность антагонистов PPARγ, препараты этой группы повышают риск инфаркта миокарда, что ограничивает их применение в терапии инсулинорезистентности.
ГПП-1, представитель класса инкретинов, способствует повышению секреции инсулина в зависимости от уровня глюкозы. В терапии СД широко применяются ингибиторы дипептидилпептидазы (глиптины), агонисты (эксенатид и лираглутид) и миметики ГПП-1. Как и метформин, миметики ГПП-1 способны проникать через ГЭБ и стимулировать соответствующие рецепторы в мозге. Эксенатид и лираглутид оказывают защитное действие на клетки мозга, подавляют нейродегенеративные процессы и предотвращают прогрессирование АД (283, 284, 285). Кроме того, наряду со снижением уровня олигомеров Aβ и нейритных бляшек, агонисты ГПП-1 стимулируют нейрогенез и улучшают распознавание объектов и пространственную память (283, 286, 287) и эффективны в терапии деменции, независимо от наличия СД.
Интраназальное введение инсулина способствует эффективному проникновению гормона в ЦНС и улучшению состояния пациентов с умеренными когнитивными расстройствами и СД2 (240, 288–290, 291). Дополнительной мишенью в терапии инсулинорезистентности служит недостаточность инсулиновой сигнализации в результате фосфорилирования серина и треонина IRS-1 (292).



