Синдром ЕЕС. Возможности пренатальной диагностики и особенности медико-генетического консультирования

УЗИ сканер HS70
Точная и уверенная диагностика. Многофункциональная ультразвуковая система для проведения исследований с экспертной диагностической точностью.
Введение
Манифестные признаки синдрома ЕЕС (синдромальное ядро)
Эктодермальная дисплазия проявляется особенностями строения всех производных эктодермы. Изменения затрагивают волосы, зубы, ногти [11]. Волосы и ресницы у таких людей тусклые, редкие и жесткие. Характерны аномалии зубов, гипоплазия зубной эмали, снижение пигментации волос, кожи, радужной оболочки глаз, что вызывает светлый их цвет. Также часто сочетание синдрома с фотобоязнью [1], обструкцией носо-слезных каналов [12], кондуктив- ной тугоухостью [13].
Эктодермальная дисплазия проявляется отсутствием потовых желез, что характеризуется наличием гипогидротинового синдрома, проявляющегося в резкой сухости и шелушении кожи. Характерным проявлением этого синдрома является хриплый, осипший голос из-за нарушения увлажнения голосовых связок, вследствие чего не происходит полного их смыкания [14].
Нарушение речи у больных с ЕЕС-синдромом имеет много причин. Во-первых, это связано с последствиями наличия расщелины губы и/или неба и «гиперназальной» речью, во-вторых, правильной артикуляции может препятствовать не только изменение голосовых связок, но и патология зубов. В-третьих, из-за аномалий слуховых косточек часто прогрессирует тугоухость, ведущая к когнитивным расстройствам и расстройствам речи.
Дети с этим синдромом обычно имеют нормальный уровень интеллекта. Лечение основано на коррекции пороков лица и конечностей. Признаки эктодермальной дисплазии проявляются в постнатальном периоде [1]. Врожденные пороки развития (ВПР) мочевыделительной системы у больных с синдромом ЕЕС возникают более чем в половине случаев. Среди них описаны: мегауретер, гидронефроз, уретероцеле, аплазия почки, аномалии гениталий, уретровезикальный рефлюкс с частыми инфекциями мочевыделительных путей [12].
Пренатальная диагностика синдрома ЕЕС
Учитывая выраженность проявлений при синдроме ЕЕС, пренатальная диагностика этой патологии, безусловно, возможна. Однако на сегодняшний день в мировой литературе встречается незначительное число публикаций, посвященных диагностике этого редкого синдрома 15, что связано, видимо, с редкостью возникновения данной аномалии. В основном сроками постановки диагноза являются 16-30 недель беременности. Большую помощь в диагностике данной патологии, учитывая выраженные изменения фенотипа лица и конечностей, оказывает применение новых ультразвуковых технологий 3D/4D с методиками поверхностной реконструкции [16].
Первое описание пренатальной диагностики синдрома датировано 1993 годом и принадлежит M. Bronshtein и R. Gershoni, когда при применении трансвагинальной эхографии у плода была выявлена расщелина губы и неба и эктродактилия в 14 недель беременности [17]. Уникальность этого факта в том, что, пожалуй, впервые из известных случаев диагностики врожденных аномалий дебют дородового выявления порока принадлежит сроку первого триместра. Примечательно и то, что авторами не только описаны отдельно выявленные пороки развития плода, но и пренатально установлена нозология этого состояния. Другими словами, диагноз не звучал, как «множественные врожденные пороки развития», когда невозможно говорить об этиологии заболевания, а следовательно, и о мерах специфической профилактики данной патологии в дальнейшем в семье. Пренатальный диагноз был выставлен полно и абсолютно корректно, что особенно важно при проведении медико-генетического консультирования с формированием тактики адекватного репродуктивного поведения семьи в дальнейшем.
Из-за незначительного количества публикаций, посвященных пренатальной диагностике этого генетического синдрома, представляем ряд собственных наблюдений диагностики синдрома ЕЕС в разные сроки беременности, в том числе и в первом триместре. Особо рассмотрим особенности проведения медико-генетического консультирования при диагностике различных форм синдрома с аутосомно-доминантным типом наследования для выработки специфических мер профилактики данного наследственного заболевания.
Клиническое наблюдение 1
Пациентка Н. 24 лет. Беременность первая. Женщина и муж соматически здоровы, брак неродственный. Обратилась в медико-генетическое отделение МОНИИАГ в 21,4 недели беременности в связи с подозрением на порок развития конечностей у плода. Были выявлены: эктродактилия кистей (рис. 1) и стоп плода (рис. 2.). Из особенностей строения выявлены двусторонние пиелоэктазии (рис. 3). Проведено медико-генетическое консультирование. Семья приняла решение прервать данную беременность в связи с наличием инвалидизирующих пороков конечностей. При патологоанатомическом исследовании ВПР конечностей подтверждены, а также дополнительно выявлена расщелина мягкого неба и признаки эктодермальной дисплазии (гипертрофия десен, характерный лицевой фенотип). Окончательный диагноз: «синдром ЕЕС, аутосомно-доминантный тип наследования». Мутация de novo.
Научная электронная библиотека

Воинова В. Ю., Юров И. Ю., Ворсанова С. Г., Юров Ю. Б.,
Классификация Х-сцепленной умственной отсталости
С клинической точки зрения Х-сцепленную умственную отсталость принято разделять на синдромальную (MRXS – mental retardation, X-linked, syndromic) и несиндромальную (неспецифическую, MRX – mental retardation, X-linked) [Kerr et al., 1991]. При синдромальных формах Х-сцепленной умственной отсталости помимо нарушений интеллекта обнаруживают различные аномалии при клиническом осмотре, лабораторных и функциональных исследованиях. Больные могут иметь нарушения физического развития, пороки развития мозга и других органов, комплекс микроаномалий, неврологические симптомы, особенности поведения, метаболические нарушения. При несиндромальных формах эти признаки отсутствуют, и наблюдается изолированная умственная отсталость [Chiurazzi et al., 2004]. Деление на синдромальные и несиндромальные формы является общепринятым, обоснованным с клинических позиций, и при этом не является специфичным только для Х-сцепленной умственной отсталости, а используется в отношении всех, в том числе аутосомных форм.
Суммарный состав моногенных состояний, классифицируемых как Х-сцепленная умственная отсталость, включает более 200 нозологических единиц [Chiurazzi et al., 2008]. По данным Генетического центра Гринвуда [Greenwood Genetic Centre site, 2016] из 126 идентифицированных
генов Х-сцепленной умственной отсталости 105 генов связаны с синдромальными её формами. Поиск генетических причин несиндромальных форм XLMR связан со значительными трудностями, поскольку при MRX отсутствуют другие клинические критерии диагностики, кроме снижения IQ и Х-сцепленного характера родословной. К настоящему времени идентифицировано 50 генов MRX [Greenwood Genetic Centre site, 2016]. Сложности дифференциальной диагностики синдромальных и несиндромальных Х-сцепленных состояний с точки зрения этиологии связаны с тем, что в обеих группах заболеваний были обнаружены мутации в одних и тех же генах [Kleefstra, Hamel, 2005]. Одновременно с синдромальной и несиндромальной XLMR связаны 29 генов. Например, мутации гена RSK2 встречаются как при синдроме Коффина – Лоури (синдромальная форма ХLMR), так и при неспецифической Х-сцепленной умственной отсталости. Мутации гена МEСР2 могут вызывать у лиц женского пола как классическую форму RTT, атипичные формы заболевания, так и несиндромальную умственную отсталость с различной степенью снижения интеллекта. Возможно, что тип мутации, локализация её в различных доменах соответствующих белков либо генетическое окружение могут влиять на фенотип, приводя к синдромальной или несиндромальной формам заболевания.
Поскольку синдромальная Х-сцепленная умственная отсталость характеризуется аномалиями различных органов и систем, рядом авторов предпринята её клиническая классификация. В частности, Chiurazzi с соавторами [2004] предложено деление синдромальной ХLMR на следующие классы:
1) синдромы мальформаций, характеризующиеся множественными врожденными аномалиями,
2) нервно-мышечные заболевания, в основном проявляющиеся неврологическими симптомами и патологией мышечной системы (атаксия, гиперкинезы, нарушения мышечного тонуса и др.),
3) метаболические болезни, возникающие вследствие дефицита определенных ферментов,
4) доминантные состояния, при которых заболевание наблюдается у девочек, а мальчики погибают внутриутробно [Chiurazzi et al., 2004].
Однако, начиная с 2008 г., от подобного деления было решено отказаться, поскольку в нем смешивались сразу несколько принципов
классификации (особенности наследования, наличие мальформаций, метаболических расстройств и др.). Так, в каталоге Chiurazzi [2008] MRXS разделена лишь на 2 класса: синдромы с мальформациями и нервно-мышечные болезни, что, с нашей точки зрения, представляется
не совсем оправданным. Так, синдром RTT, согласно делению Chiurazzi, был отнесен к нервно-мышечным заболеваниям, под которыми авторы подразумевают синдромы, характеризующиеся умственной отсталостью и неврологическими симптомами (эпилепсией, спастической параплегией, атаксией и др.). Однако данная классификация вступает в противоречие c общепринятым понятием «нервно-мышечные заболевания», подразумевающим группу болезней, связанных с нарушением функции произвольной мускулатуры вследствие поражения собственно мышц, нервно-мышечных соединений, периферических нервов или моторных нейронов спинного мозга [Гехт, 1982]. Следует отметить, что RTT проявляется, прежде всего, психическими, но не мышечными нарушениями, и поэтому согласно международной классификации болезней (МКБ-10) относится к разделу «Расстройства психологического развития» класса «Психические расстройства и расстройства поведения».
По нашему мнению, помимо обоснованного разделения на синдромальную и несиндромальную Х-сцепленную умственную отсталость, удобной является классификация, основанная на характере наследования заболевания. Среди всех форм XLMR преобладают те, которые проявляются преимущественно у гемизигот по Х-сцепленным мутациям. В настоящее время выделено менее десятка так называемых доминантных состояний, проявляющихся преимущественно у гетерозигот. Однако они заслуживают отдельного рассмотрения, так как в силу высоких частот ряда нозологических форм вносят значительный вклад в XLMR (табл. 2).
Доминантные формы Х-сцепленной умственной отсталости
Журнал «Здоровье ребенка» 5(20) 2009
Вернуться к номеру
Алгоритм диагностики основных наследственных синдромов, проявляющихся аномалией развития сердца и нервной системы
Авторы: Евтушенко С.К., Морозова Т.М., Прохорова Л.М., Голубева И.Н., Перепечаенко Ю.М., Зима И.Е., Евтушенко Л.Ф., Лепихов П.А.; Донецкий национальный медицинский университет им. М. Горького, Областная детская клиническая больница, г. Донецк
Версия для печати
Синдромальные формы патологии составляют значительную часть интеграционных болезней в медицине. Мы представляем алгоритм клинической диагностики наследственных синдромов, ядром которого являются врожденные аномалии сердца в сочетании с патологией как нервной системы, так и других органов и систем.
Синдромология в педиатрии — чрезвычайно обширная область, охватывающая практически все медицинские специальности. Несмотря на то что некоторые синдромы достаточно редки, суммарно синдромальные формы патологии составляют существенную часть интеграционных болезней в медицине. Около 6 % всех новорожденных имеют множественные врожденные аномалии, а в 40 % случаев из них уже сегодня диагностированы определенные хромосомные и генетические синдромы. В той или иной степени синдромальной патологией у детей занимаются педиатры, кардиологи, ортопеды, неврологи, отоларингологи, окулисты и др. Но врачи первичного звена медицинской помощи (педиатры, семейные врачи) часто оказываются первыми, к кому обращаются пациенты с сочетанной, в том числе и неврологической симптоматикой. На патологию сердца, легких или нервной системы врач обращает внимание еще в младенческий период. Но сложность состоит в том, что каждый специалист видит прежде всего «свою» патологию, хотя чаще доминирует патология нервной системы и сердца.
Детская неврология как наука за последние десятилетия значительно изменилась. Это связано с внедрением новых медицинских технологий, в первую очередь лучевой диагностики, возросли поток современной медицинской информации и число лекарственных препаратов. Однако в связи с реформированием практического здравоохранения в последние 5 лет возник дефицит детских неврологов. На этом фоне знания детской амбулаторной неврологии у врачей педиатрического профиля остаются недостаточными. Предусмотренный интернатурой модуль на кафедре детской неврологии длится 3 недели, что крайне недостаточно, а пятилетний цикл переаттестации не удовлетворяет потребности педиатров в дальнейшем получении оперативной медицинской информации. Все чаще стали допускаться со стороны врачей первичного звена диагностические ошибки. К сожалению, и семейные врачи, и педиатры сегодня не могут оценить неврологический статус ребенка.
Мы представляем алгоритм клинической диагностики наследственных синдромов, ядром которого являются врожденные аномалии сердца в сочетании преимущественно с патологией нервной системы.
Без сомнения, проблема междисциплинарная. В настоящее время особенно она касается педиатров-кардиологов, детских неврологов и кардиохирургов. Сегодня оперативное вмешательство по поводу пороков сердца у младенцев стало действительностью. Нам представляется, что в сферу наблюдения кардиологов и кардиохирургов чаще попадают дети не с наследственными синдромами, а с сочетанными пороками сердца и аномалиями развития мозга (микрогирия, гипоплазия, агенезия мозолистого тела и др.), и они встречаются чаще. Подобные феномены в тактике наблюдения, особенно хирурга, должны учитываться обязательно!
2009/3_help/help_4_1.jpg)
2009/3_help/help_4_2.jpg)
2009/3_help/help_4_3.jpg)
2009/3_help/help_4_4.jpg)
2009/3_help/help_4_5.jpg)
2009/3_help/help_4_6.jpg)
Основной задачей думающего врача является не только применение современных методов исследования. Важно учитывать весь клинический спектр патологических симптомов и синдромов у каждого конкретного ребенка.
Мы надеемся, что предложенный алгоритм клинической диагностики поможет практическому врачу правильно осмыслить полученные результаты объективного и инструментального обследования и установить диагноз пациентам с сочетанной аномалией развития сердца и нервной системы, а также других органов и систем.
1. Вельтищев Ю.Е., Казанцева Л.З., Семячкина А.Н. Наследственные болезни обмена веществ // Наследственная патология человека / Под ред. Ю.Е. Вельтищева, Н.П. Бочкова. — М., 1992.
2. Вельтищев Ю.Е., Темин П.А. Наследственные болезни нервной системы. — М.: Медицина, 1998.
3. Евтушенко С.К., Морозова Т.М., Шестова Е.П., Евтушенко О.С. Синдром мышечной гипотонии у новорожденных и детей раннего возраста. Учебно-методическое пособие. — Донецк, 2008.
4. Скворцов И.А., Ермоленко Н.А. Развитие нервной системы у детей в норме и патологии. — М.: МЕДпресс-информ, 2003.
5. Феничел Дж.М. Педиатрическая неврология: Основы клинической диагностики: Пер. с англ. — М.: ОАО «Медицина», 2004.
Современные подходы к изучению плодового фенотипа в эру генетического ультразвука

УЗИ аппарат HS40
Лидер продаж в высоком классе. Монитор 21,5″ высокой четкости, расширенный кардио пакет (Strain+, Stress Echo), экспертные возможности для 3D УЗИ в акушерско-гинекологической практике (STIC, Crystal Vue, 5D Follicle), датчики высокой плотности.
Настоящая работа посвящена изучению плодового фенотипа при ультразвуковом исследовании в различные сроки беременности при помощи новейших технологий Samsung, оптимизирующих диагностику многих врожденных пороков развития, аномалий, нарушений строения, особенностей развития с оценкой значимости отдельных ультразвуковых патологических маркеров в качестве потенциальных признаков генетических синдромов и ассоциаций.
Одной из ведущих причин детской и младенческой смертности являются врожденные пороки развития (ВПР), среди которых в 20% случаев встречаются множественные ВПР (МВПР) с установленной частотой 1:250 новорожденных [1]. Особое значение в профилактике МВПР имеет пренатальная диагностика – современный и высокоэффективный метод диспансеризации плода, направленный на своевременное выявление патологии, впоследствии определяющий выбор адекватной акушерской тактики при данной беременности и специфических мер профилактики болезни в данной семье в дальнейшем [2]. Современные ее возможности позволяют из комплекса патологических нарушений у плода выделить достоверно значимые ультразвуковые маркеры для постановки диагноза генетических синдромов различной этиологии. Сегодня пренатальная синдромология активно развивается, как в плане расширения спектра возможных лабораторных исследований, так и описания значимых, ключевых, таргетных ультразвуковых признаков многих наследственных синдромов различного происхождения.
Группа МВПР включает сотни нозологических форм, различных по этиологии, фенотипическим проявлениям и прогнозу. В силу огромной гетерогенности МВПР представляют собой сложную проблему для постановки окончательного диагноза. Основными этиологическими факторами, приводящими к возникновению синдромов с множественными пороками развития, являются генные, хромосомные мутации и действие на плод неблагоприятных факторов внешней среды [3, 4].
Любые наследственные или врожденные генетические болезни являются результатом повреждения генетической информации на геномном, хромосомном или генном уровне. В 60% всех случаев МВПР встречаются хромосомные синдромы, достаточно широко изученные и хорошо диагностируемые. Для лабораторной диагностики этих повреждений разработаны разнообразные и хорошо известные методы: цитогенетический, молекулярно-цитогенетический и молекулярно-генетический. МВПР нехромосомного генеза встречаются в 40% случаев. Среди них выделяют синдромы, ассоциации и неклассифицированные комплексы. Применительно к нехромосомным синдромам цитогенетическое исследование позволяет лишь отвергнуть хромосомную природу данного комплекса пороков развития, но не установить диагноз.
Синдромы с известным типом наследования представлены моногенными синдромами, в структуре которых наиболее часто встречаются аутосомно-доминантные (АД) и аутосомно-рецессивные (АР) нозологии, и спорадическими синдромами с различными известными ассоциациями (устойчивыми сочетаниями ВПР с этиологической общностью). Неклас си фицированные комплексы МВПР в международном классификаторе наследственных синдромов и ассоциаций (OMIM- www. omim.org) не зарегистрированы, судить о типе их наследования не представляется возможным в связи с отсутствием опубликованных данных о причинах их возникновения. Большая часть неклассифицированных комплексов представляет собой случайное сочетание 2–3 ВПР, однако некоторые из них могут быть не случайны, а пред ставлять собой новые, не описанные и не выделенные в самостоятельные нозологические формы синдромы [5].
Сегодня имеется определенное количество публикаций о выявлении некоторых синдромов и ассоциаций при пренатальной эхографии [6, 7]. Однако дородовая идентификация нозологических форм нехромосомных синдромов пока не вошла в рутинную практику врачей в области пренатальной диагностики и носит случайный характер. В литературе отсутствуют четкие рекомендации о системном подходе к диагностике наследственных синдромов различной этиологии, нет диагностических алгоритмов и определенных схем взаимодействия врачей ультразвуковой пренатальной диагностики, генетиков, акушеров-гинекологов.
Использование эхографии делает возможным пренатальное выявление не менее 80% врожденных дефектов во II, III триместрах беременности и свыше 50% значимых нарушений анатомии плода уже в I триместре начиная со сроков 11–12 нед. Весь комплекс диагностированных пороков развития и различных микроаномалий в совокупности составляет фенотипический спектр синдрома, который состоит из «ядерных» (главных, ключевых) признаков и дополнительных аномалий, наличие которых не является обязательным для диагностики клинической нозологии синдрома.
Революцией в пренатальной ультразвуковой диагностике явилось появление объемной эхографии, которая, обладая такими качествами, как неинвазивность, безопасность и возможность многократного применения у одной пациентки, имеет высокую информативность в исследовании анатомии плода и изучении его фенотипа. При применении различных режимов объемной эхографии абсолютно очевидно их преимущество по сравнению с обычным сканированием. Детально можно изучить лицо плода (рис. 1–4) в различные сроки беременности, начиная со сроков первого пренатального скрининга в 11–14 нед, конечности плода, причем не только их наличие и положение (рис. 5, 6), но и состояние и количество пальцев (рис. 7–9) как на руках, так и на ногах. Также можно изучить позвонки плода (рис. 10), состояние твердого нёба (рис. 11, 12), строение наружного уха (ушной раковины) (рис. 13), состояние основных швов черепа и родничков, исключая их преждевременное закрытие при кранисиностозах (рис. 14, 15).
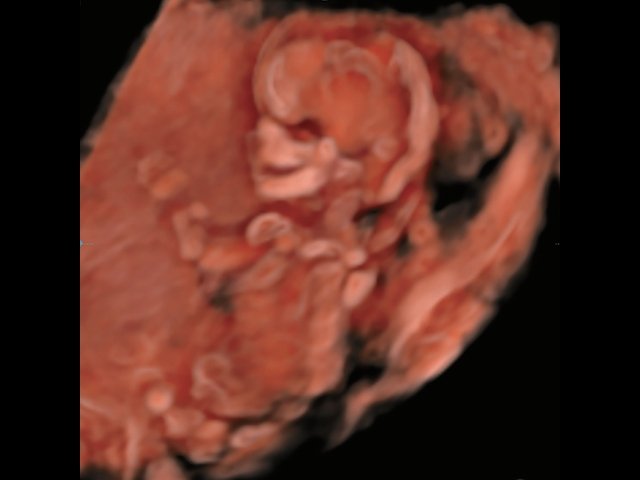
Рис. 1. Нормальное лицо плода, 12 нед беременности.
Классификация, диагностика и лечение идиопатической низкорослости
Полный текст:
Аннотация
Ключевые слова
Для цитирования:
Шандин А.Н., Петеркова В.А. Классификация, диагностика и лечение идиопатической низкорослости. Проблемы Эндокринологии. 2009;55(4):36-44. https://doi.org/10.14341/probl200955436-44
For citation:
Shandin A.N., Peterkova V.A. Classification, diagnosis, and treatment of idiopathic short stature. Problems of Endocrinology. 2009;55(4):36-44. (In Russ.) https://doi.org/10.14341/probl200955436-44
Рост является одним из главных показателей здоровья и благополучия ребенка. Жалобы на низкий рост — одна из частых причин обращаемости к детскому эндокринологу.
Ребенок считается низкорослым, если его рост более чем на 2 SDS ниже среднего роста детей его возраста и пола в популяции. Причинами этого могут быть хронические системные заболевания, эндокринопатии, недостаточность питания, хромосомные нарушения и др. (табл. 1).
В то же время, несмотря на расширяющиеся диагностические возможности, в 60—80% случаев причину отставания определить так и не удается, и такую низкорослость называют идиопатической [7].
Таким образом, идиопатическая низкорослость представляет собой гетерогенную группу состояний, включающих в себя как нераспознанную патологию, так и варианты нормального развития (конституциональная задержка роста). Понятно, что они имеют разный ростовой прогноз. С учетом этого усилия врача должны быть направлены на поиск возможных причин и выбор оптимальной тактики ведения таких детей.
Проблема диагностики и лечения идиопатической низкорослости является предметом постоянных споров и дискуссий. В последнее время с расширением показаний к терапии гормоном роста (ГР) при состояниях, не сопровождающихся дефицитом соматотропного гормона (СТГ), остро встал вопрос о его применении при идиопатической низкорослости. К настоящему времени в мире накоплено много данных об эффективности и безопасности лечения ГР детей с идиопатической низкорослостью. В 2008 г. был опубликован Международный консенсус по диагностике и тактике ведения идиопатической низкорослости, созданный в результате совместной работы трех научных обществ: Общества по изучению гормона роста, Общества педиатров-эндокринологов им. Лоусона Вилкинса (США) и Европейского общества педиатров-эндокринологов [4]. Основные положения данного документа были одобрены другими международными обществами детских эндокринологов (Латиноамериканским, Японским, Канадским, Азиатским и Австралийским).
Классификация
Важным моментом для постановки диагноза и выбора тактики ведения больных является терминология и классификация. Ниже представлены старая и новая классификации идиопатической низкорослости.
Классификации идиопатической низкорослости
а) семейная низкорослость;
б) конституциональная задержка роста и пубертата (КЗРП);
в) сочетание семейной низкорослости и КЗРП. Классификация ESPE (2007):
а) семейная низкорослость:
б) несемейная низкорослость:
Ранее использовавшаяся классификация идиопатической низкорослости, широко распространенная в России, в мире к настоящему времени практически не применяется. Это связано с несколькими объективными моментами. Диагноз КЗРП можно поставить не ранее возраста 13—14 лет. До этого момента точно сказать, является низкий рост ребенка вариантом нормы (конституциональная задержка роста) или патологией (несемейная форма низкорослости), невозможно. Применение в качестве критерия конституциональной задержки роста степени задержки костного возраста неоправданно, так как отставание костного возраста не всегда эквивалентно отставанию в сроках пубертата [11]. Некоторые дети с задержкой костного созревания вступают в пубертат вовремя и наоборот [12].
Семейная низкорослость в широком понимании представляет собой все формы низкорослости с отягощенным семейным анамнезом, т. е. она включает в себя и семейные формы СТГ-дефицита, и синдромальную (синдром Нунан), и костную патологию. В узком понимании под семейной низкорослостью понимают собственно идиопатическую низкорослость с отягощенным семейным анамнезом.
Использование в качестве критерия семейной низкорослости соответствия костного возраста хронологическому неправильно, так как не отражает действительности. При семейной низкорослости костный возраст очень часто отстает от паспортного. Соответствие костного и хронологического возраста более характерно для семейных случаев костных дисплазий.
Таким образом, старая классификация охватывает фактически лишь детей с предполагаемой семейной костной патологией и подростков с КЗРП. В то же время большая группа детей с нормальным выбросом СТГ на стимуляции оказывается неохваченной. Некоторые авторы вообще предлагают не подразделять идиопатическую низкорослость на подгруппы, ссылаясь на отсутствие четких критериев, позволяющих ясно дифференцировать эти два состояния [24]. Однако большинство эндокринологов, и это отражено в Международном консенсусе 2008 г., все же высказываются о необходимости классификации идиопатической низкорослости на подгруппы.
Таблица 1. Причины отставания в росте
Внутриутробная задержка роста
Хронические системные заболевания и пороки развития:
Психоэмоциональные и стрессорные факторы
Недостаточность питания, голодание Ятрогенная
Задержка внутриутробного развития
Синдромы Шерешевского—Тернера, Нунан, Сильвера—Рассела, Секкеля и др. Костные дисплазии
Бронхиальная астма, муковисцидоз
Хроническая почечная недостаточность, нефрогенный несахарный диабет
Любая патология органа
Болезнь Крона, целиакия, мальабсорбция
Гликогенозы, мукополисахаридозы, липидозы
Депривационная, стрессорная задержка роста
Нервная анорексия, задержка роста на фоне недостаточного питания Задержка роста на фоне облучения, приема лекарственных препаратов СТГ-дефицит, гипотиреоз, синдром Кушинга, гипогонадизм, сахарный диабет
По современной классификации, принятой ESPE в 2007 г. и одобренной к применению Международным консенсусом 2008 г., выделяют две формы идиопатической низкорослости — семейную и несемейную [23]. В качестве отличительного критерия используют рост родителей. Для этого рассчитывается целевой (генетический) рост по формуле Tanner:
Рост отца + рост матери ± 13 см.
Границы целевого роста ± 10 см. При семейной форме идиопатической низкорослости ребенок низкорослый по сравнению с общей популяцией, но в то же время его рост остается в пределах целевого роста для семьи (SDS роста ребенка в пределах SDS целевого роста ± 1,5 SD). При несемейной форме ребенок низкорослый как для общей популяции, так и для своей семьи (рис. 1). Кроме того, семейную низкорослость можно диагностировать даже при соответствии роста ребенка целевому росту в случаев наличия других низкорослых родственников (один родитель, бабушки, дедушки). Это связано с тем, что генетический дефект может передаваться рецессивно или доминантно-негативно.
Низкорослыми в данном случае считаются родственники с ростом менее —1,5 SDS, т. е. мужчины ниже 165 см, женщины ниже 154 см.
У детей старше 13—14 лет обе формы подразделяют на низкорослость с задержкой пубертата и без нее. Задержка пубертата ставится у девочек в 13 лет при отсутствии увеличения молочных желез, у мальчиков в 14 лет — при объеме яичек меньше 4 мл. Несемейную низкорослость с задержкой пубертата также называют КЗРП.
Диагностика
Идиопатическая низкорослость (ИН) — диагноз исключения. Его ставят, когда все возможные диагнозы были исключены. Понятно, что все зависит от диагностических возможностей врача (возможность проведения СТГ-стимуляционных проб, теста на генерацию инсулиноподобного фактора роста (ИФР), молекулярногенетического анализа). Так или иначе, после установления какого-либо конкретного диагноза диагноз «идиопатическая низкорослость» более не применяют.
Алгоритм диагностики идиопатической низкорослости представлен на рис. 2.
Анамнез. Оценка любого ребенка с низким ростом всегда должна начинаться с тщательного сбора анамнеза (анамнез жизни, семейный анамнез, анамнез заболевания) и расширенного физикального осмотра (с оценкой фенотипа, пропорций тела, полового и психологического статуса).
Правильно собранный анамнез еще до проведения лабораторно-инструментальных исследований позволяет исключить внутриутробную задержку роста, хронические системные заболевания и пороки развития органов дыхания, сердечно-сосудистой системы, крови, печени, почек, желудочно-кишечного тракта, которые могут быть причинами низкорослости.
Семейный анамнез необходим для классификации. Обязательно строят родословное древо, указывают национальность, наличие близкородственного брака. Рост родителей (желательно не со слов, а измеренный врачом лично) необходим для вычисления целевого роста. Кроме того, оценивают их пропорциональность (для исключения костных дисплазий), а также ищут похожий фенотип (например, характерные черты лица при синдроме Нунан). Для этого может потребоваться изучение семейных фотографий.
Помимо этого, выясняется время начала пубертата и динамика роста у родителей и родственников (менархе у женщин, появление оволосения, ростовой скачок, пролонгированный рост у мужчин). Часто при конституциональной задержке роста у родителей (как правило, у мужчин) наблюдается аналогичный вариант развития: низкий рост с 2 лет чуть ниже 3-й перцентили на протяжении всего детства, позднее (с задержкой на 2—4 года) начало пубертата и поздний ростовой скачок с достижением нормального конечного роста.
Антропометрия. Важным моментом в процессе диагностики является сравнение роста ребенка с возрастными нормами, с вычислением антропометрических параметров (SDS роста, скорость роста, SDS скорости роста) и обязательным построением кривых роста. Применение перцентильных кривых необходимо при диагностике, но особенно важно в процессе наблюдения (для выбора тактики ведения) и лечения (для оценки эффективности терапии) таких детей. Некоторые типы кривых очень характерны для определенных заболеваний и иногда позволяют поставить диагноз (КЗРП, краниофарингиома, недостаточность питания, гиперкортицизм). В современной практике все основные показатели, применяемые детскими эндокринологами (хронологический возраст, SDS роста, SDS скорости роста и многие другие) рассчитываются с помощью специальных электронных программ (Auxology, Growthanalyser).
Клинический осмотр включает в себя, помимо общетерапевтического осмотра, оценку фенотипа, пропорций тела, полового и психологического статуса. При тщательном осмотре ребенка может быть выявлен ряд синдромов и заболеваний, сочетающихся с низкорослостью (синдромы Шерешевского—Тернера, Нунан, Сильвера- Рассела, Секкеля; врожденный гипотиреоз, гиперкорти- цизм и ряд других).
Измерение роста сидя с оценкой пропорциональности необходимо для исключения костных дисплазий (ги- похондроплазия, спондилоэпифизарная дисплазия). При наличии диспропорции необходимы специальные рентгенологические исследования, консультация генетика и специалиста по костной патологии. В некоторых случаях возможно молекулярно-генетическое подтверждение диагноза (например, идентификация мутаций гена FGFR3 при гипохондроплазии).
Оценка полового статуса (по Таннеру) позволяет определить отставание в половом развитии. Задержка пубертата при отставании в росте может быть как конституциональной (при КЗРП), так и патологической (при гипогонадизме). При последнем необходимо специфическое обследование (проба с бусерелином, измерение Л Г, ФСГ и половых стероидов и т. д.).
Помимо этого, важно отметить наличие правильного строения гениталий. При неправильном строении гениталий у мальчика возможна дисгенезия гонад и требуется исследование кариотипа. При дисгенезиях, как у мальчиков, так и у девочек, наблюдается отставание в росте.
При сборе анамнеза и осмотре важно оценить отношение родителей и самого ребенка к проблеме. Оценивается социальная адаптация (черты характера, успеваемость в школе, интересы).
Лабораторная диагностика
Рекомендуемые лабораторные исследования при идиопатической низкорослости представлены в табл. 2. Кроме того, в процессе лечения исследуют уровни глюкозы, иммунореактивного инсулина (ИРИ), гликированного гемоглобина (НЬА1С) (для контроля углеводного обмена).
Кариотипирование. Проводится всем девочкам с отставанием в росте, мальчикам с неправильным строением наружных гениталий и при подозрении на хромосомные синдромы.
СТ Г-стимуляционные тесты. Золотым стандартом для исключения СТГ-дефицита во всем мире является проведение стимуляционных тестов. В настоящее время используется множество разных тестов. Но ни один из них, ни даже их сочетание не являются абсолютно надежным критерием из-за недостаточной специфичности, получения ложноотрицательных результатов и дискондартности разных проб.
Традиционно (согласно Российскому консенсусу 2005 г., а также по Международному и национальным консенсусам разных стран) нормальным выбросом СТГ на стимуляции считается выброс выше 10 нг/мл [2]. Диагноз «тотальный СТГ-дефицит» ставят при пике выброса менее 7 нг/мл, парциальный СТГ-дефицит — при пике выброса от 7 до 10 нг/мл. После получения пика выброса более 10 нг/мл необходимости в проведении второй пробы нет, диагноз СТГ-дефицита исключен. В то же время при отсутствии множественного дефицита гормонов аденогипофиза, MP-картины «триады» (гипоплазия гипофиза, аплазия ножки гипофиза, эктопия нейрогипофиза) или сочетания выраженного отставания в росте (SDS 0,3—0,5 через 1 год терапии;
— увеличении скорости роста более чем на 3 см/год;
— SDS скорости роста больше +1.
Плохой ростовой ответ может быть следствием либо плохой комплаентности, либо нечувствительности к ГР. При хорошей комплаентности нужно еще раз пересмотреть диагноз и проверить другие возможные причины отставания в росте. При подозрении на резистентность к ГР следует провести тест на генерацию ИФР-1 на фоне отмены терапии, если его не проводили до начала лечения. После этого можно либо увеличить дозу, либо прекратить лечение. При хорошем ростовом ответе лечение продолжают, не меняя дозу ГР.
Продолжительность терапии
Существуют две точки зрения на этот счет. Согласно первой, лечение продолжают до достижения роста, близкого к конечному (до закрытия зон роста); скорость роста менее 2 см/год и/или костный возраст старше 16 лет у мальчиков и старше 14 лет у девочек. Согласно другой точке зрения, терапию нужно прекращать уже при достижении «нормального» роста. Обычно это рост выше — 2 SDS, но можно использовать и другие значения (10-я перцентиль или даже 50-я перцентиль). Лечение можно прекратить и раньше, если ребенок или его родители удовлетворены достигнутым ростом или не желают продолжать терапию по другим причинам.
Возможные побочные эффекты
По данным многочисленных контролируемых исследований, частота побочных эффектов при терапии ГР детей с идиопатической низкорослостью не выше, чем при терапии ГР других состояний [10]. Однако, учитывая возможный риск возникновения нарушений углеводного обмена и опухолей, необходим тщательный мониторинг.
Параметры, контролируемые на фоне лечения ГР, приведены в табл. 4.
Проблемы лечения идиопатической низкорослости гормоном роста
Главная проблема в терапии идиопатической низкорослости — это ее высокая стоимость. Нет сомнений, что биосинтетический ГР — дорогое удовольствие. Ежегодная цена лечения одного ребенка 10 лет, массой тела 20 кг, составляет в среднем 200—300 тыс. рублей. Общая сумма, затраченная на лечение такого ребенка до достижения им конечного роста, при сроке терапии 5—7 лет теоретически составляет около 1 — 1,5 млн руб. И тогда цена 1 ’’лишнего» сантиметра роста становится равной 150—200 тыс. руб. Однако надо отметить, что достижение нормального роста часто происходит уже в детстве, и при этом возможно прекращение терапии. Кроме того, не совсем этично измерять здоровье и счастье человека, а особенно ребенка, в денежном эквиваленте.
Таблица 4. Мониторинг эффективности и безопасности лечения ГР



