Болезнь Хаммена-Рича
1. Что представляет собой редкая болезнь Хаммена-Рича
Из всех наук медицина, как известно – самая консервативная (добавим: к счастью для пациентов). Однако и в медицине прогресс идет достаточно быстрыми темпами; революционные открытия и прорывы сочетаются с традиционным, эволюционным накоплением знаний и методов. Заболевания, ранее совершенно непонятные, редкие и, казалось бы, беспричинные, постепенно открывают свои тайны, и в этом непрерывном процессе возникает масса новых вопросов. Примером может послужить болезнь (в других источниках – синдром) Хаммена-Рича, известная также как идиопатический фиброзирующий альвеолит (ИФА), интерстициальный пневмофиброз и т.д.
Данное заболевание названо в честь ученых, которые в 30-40 гг ХХ века впервые дали полноценное клиническое описание специфического фиброза – вытеснения функциональной ткани легких разрастающейся соединительной тканью, – вследствие которого больные погибали в сроки от одного месяца до полугода. И хотя первые описания базировались на мизерном, с точки зрения современной статистики, объеме наблюдений, – всего четыре случая, – публикации Л.Хаммена и А.Рича не прошли незамеченными. В 60-е годы значительный вклад в изучение проблемы внес Дж.Скеддинг; предложенный им термин «фиброзирующий альвеолит» акцентирует, прежде всего, наличие воспалительного процесса в альвеолах, а термин «синдром Скеддинга» некоторое время употреблялся как один из синонимов болезни Хаммена-Рича. Частота встречаемости и диагностики этого заболевания, в целом, увеличивается, как увеличивается и количество связанных с ним вопросов, – которые, как говорят в науке, «нуждаются в дальнейших исследованиях».
Болезнь Хаммена-Рича объединяет, по всей вероятности, группу заболеваний с различными пусковыми механизмами и закономерностями развития. Установлено, что средний возраст манифестации превышает 50 лет (хотя случаи ИФА у детей также не являются уникальными), что мужчины болеют несколько чаще, что наличие данной болезни в разы повышает онкологический риск, и т.д. Но всего этого, безусловно, недостаточно для полного понимания патологии и разработки ее этиопатогенетического лечения. Достоверными остаются лишь тяжесть течения ИФА, неблагоприятный прогноз и высокая летальность: воспалительный фиброз легких, чем бы он ни был вызван, приводит к прогрессирующей дыхательной недостаточности.
2. Причины
На данный момент идиопатический фиброзирующий альвеолит относят к полиэтиологическим болезням, т.е. к заболеваниям многопричинной природы (что лишний раз подтверждает невеселую медицинскую присказку: «Если причин у болезни слишком много, значит, причину мы не знаем»). Уточняющий термин «идиопатический» в названии заболевания относит этиологию к сугубо индивидуальным особенностям организма. Наиболее перспективные и доказательные из гипотез, публикуемых в отношении этиопатогенеза ИФА, связывают начало процесса с аутоиммунными расстройствами, при которых защитная система начинает атаковать собственный организм. Сообщается также о возможной связи с аллергическими состояниями, интоксикацией, инфекциями (как вирусными, так и бактериальными), а также с наследственной предрасположенностью. Однако накопленные исследовательские данные пока не позволяют ни подтвердить, ни исключить эти гипотезы.
3. Симптомы и диагностика синдрома Хаммена-Рича
Практически любая легочная патология, в т.ч. ИФА, приводит к дефициту оксигенации (насыщения тканей и органов кислородом), что, в свою очередь, проявляется одышкой, слабостью, кашлем, синюшностью кожных покровов. По мере прогрессирования деформируются пальцы рук и ногти на них (синдромы «барабанных палочек» и «часовых стекол», соотв.). Кроме того, ИФА чреват рядом серьезных осложнений со стороны других органов.
Как правило, дыхательная недостаточность развивается достаточно быстро и инвалидизирует больного (буквально вынуждая снижать темп и интенсивность жизни). Достигнув тяжелой степени и при этом оставаясь без адекватного терапевтического ответа, болезнь Хаммена-Рича результирует фатально.
Различают до пяти степеней (или стадий) заболевания; на поздних этапах легкие приобретают характерную ячеистую, как у решета, форму, – за счет множества кист, – и, все больше лишаясь основной, «рабочей» ткани, утрачивают и способность выполнять свои вентиляционные и газообменные функции.
Диагностировать болезнь Хаммена-Рича на самом раннем, доклиническом этапе не удается практически никогда: больные обращаются за помощью уже с развернутой клинической картиной. Помимо традиционной аускультации (выслушивания), которая выявляет ряд характерных акустических признаков, стандартом в диагностике данной патологии являются рентгенография и биопсия.
4. Лечение и прогноз
Неэффективность антибиотиков резко снижает доказательную базу «инфекционной гипотезы» ИФА. Наиболее действенными оказываются противовоспалительные гормональные средства в больших дозах, некоторые группы витаминов и диуретиков, иммунодепрессанты, детоксицирующие средства. Критически важным фактором является своевременность начала лечения. Возможно, интенсивные исследования и напряженный поиск эффективных терапевтических схем привели к тому, что катастрофически быстрые формы фиброзирующего альвеолита постепенно уступают место хроническим и рецидивирующим типам течения.
Однако общий прогноз остается неблагоприятным. Средняя продолжительность жизни при установленном диагнозе ИФА составляет от четырех до восьми лет.
Что такое болезнь хаммена рича
Сотрудники Курского государственного медицинского университета, Научно-исследовательского клинического института педиатрии им. академика Ю.Е. Вельтищева и РНИМУ им. Н.И. Пирогова описали клинический случай синдрома Хаммана–Рича у ребенка 8 мес, основные клинические проявления и особенности диагностики синдрома.
Синдром Хаммена–Рича (идиопатический фиброзирующий альвеолит) характеризуется патологическим процессом в альвеолах и интерстициальной ткани легких неясной природы, что приводит к прогрессирующему фиброзу и сопровождается нарастающей дыхательной недостаточностью. Первые упоминания о болезни относятся к 1935 г., когда L. Hamman и А. Rich описали ряд больных с быстропрогрессирующим легочным фиброзом и дыхательной недостаточностью, приведшим в течение нескольких месяцев к летальному исходу.
В 1999, 2002 и 2013 гг. были приняты совместные соглашения Американского торакального общества и Европейского респираторного общества по интерстициальным заболеваниям легких. В этих соглашениях были выделены и дифференцированы различные формы идиопатических интерстициальных пневмоний преимущественно по морфологическим признакам. Собственно идиопатический фиброзирующий альвеолит в настоящее время рассматривается как заболевание с морфологической картиной интерстициальной пневмонии. Другие формы интерстициальных пневмоний, в их числе острая интерстициальная пневмония (синдром Хаммана–Рича), неспецифическая интерстициальная пневмония, десквамативная интерстициальная пневмония, интерстициальная болезнь с респираторным бронхиолитом и другие типы идиопатических интерстициальных пневмоний являются самостоятельными заболеваниями.
Этиология синдрома Хаммана–Рича до конца не известна. Существовавшие многие годы теории вирусного, полиэтиологического происхождения болезни не получили подтверждения с позиции доказательной медицины. Повышенная частота полиморфизмов генов, кодирующих ряд цитокинов, профибротических молекул, матриксных металлопротеиназ, которая была показана при первоначальном изучении генетического компонента идиопатического легочного фиброза, в дальнейшем также не подтвердилась.
Гены, дефекты которых обнаружены у пациентов с идиопатическим фиброзирующим альвеолитом, включают гены, связанные с теломеразами (TERT и TERC), сурфактантными белками C ( SPC) и A2 (SPA2) и белком ELMOD2. Мутации генов TERT или TERC были выявлены примерно в 18% случаев семейного легочного фиброза (среди кровных родственников) и реже – в спорадических случаях. Эти мутации приводят к укороченной длине теломеров как у пораженных индивидуумов, так и у бессимптомных носителей, а у некоторых родственников также были продемонстрированы укороченные теломеры, что может свидетельствовать об эпигенетической природе этого феномена. Кроме того, короткие теломеры в отсутствие мутаций TERT/TERC были выявлены в 25% спорадических и 37% семейных случаев идиопатического фиброзирующего альвеолита, что указывает на другие механизмы укорочения теломеров.
Общий однонуклеотидный полиморфизм в области промотора гена MUC5B (муцин 5В) был обнаружен в 38% случаев идиопатического легочного фиброза. Следует отметить, что хотя полиморфизм промотора MUC5B, по-видимому, создает повышенный риск развития идиопатического легочного фиброза, он может быть связан с улучшением выживаемости при этом заболевании. Недавно проведенные два крупных исследования по изучению генных ассоциаций подтвердили связь синдрома Хаммана–Рича с нуклеотидными вариантами TERT, TERC и MUC5B, а также другими новыми локусами (например, TOLLIP), некоторые из которых, как полагают, участвуют в клеточно-клеточной адгезии и репарации ДНК.
С учетом патогенеза предполагается, что в интерстициальной ткани легких снижается распад коллагена и повышается его синтез фибробластами и альвеолярными макрофагами. По данным A. Ferreira и соавт., заболевание относится к аутоиммунным. Комплексы антиген–антитело откладываются в стенках мелких сосудов легких. Под влиянием циркулирующих иммунных комплексов, лизосомальных ферментов альвеолярных макрофагов и нейтрофилов происходят повреждение легочной ткани, уплотнение, утолщение межальвеолярных перегородок, облитерация альвеол и капилляров фиброзной тканью. Патоморфологические изменения в легких при этом заболевании происходят в виде трех взаимосвязанных процессов: интерстициальный отек, интерстициальное воспаление (альвеолит) и интерстициальный фиброз.
В клинической картине заболевания определяющая роль принадлежит дыхательной недостаточности. Одышка – главный симптом практически у всех больных идиопатическим легочным фиброзом, наблюдается у большинства пациентов, особенно у детей младшего возраста и служит наиболее ранним признаком заболевания. Дыхательная недостаточность вначале возникает или усиливается при физической нагрузке, но неуклонно прогрессирует. У больных, как правило, отмечается кашель – непродуктивный или со скудной слизистой мокротой. Цианоз – менее постоянный и более поздний признак болезни, возникает или усиливается при физической нагрузке, у маленьких детей при кормлении.
В процессе болезни отмечается значительное похудание детей, отставание в росте. Частый и прогностически неблагоприятный признак – утолщение концевых фаланг пальцев по типу барабанных палочек, ногтей в форме часовых стекол («пальцы Гиппократа»). С большим постоянством наблюдается деформация грудной клетки.
Обнаруживаемые при физикальном обследовании изменения в легких достаточно специфичны. У больных на вдохе прослушиваются нежные крепитирующие хрипы («треск целлофана»). Они могут быть непостоянными по своей выраженности и локализации. Несоответствие выраженной одышки относительно небольшим физикальным изменениям в легких служит одним из важнейших дифференциально-диагностических признаков, позволяющих клинически отличить синдром Хаммана–Рича от других хронических заболеваний бронхолегочной системы. На поздних стадиях заболевания, как правило, отмечаются нарастание одышки, формирование легочно-сердечной недостаточности за счет гемодинамических нарушений в малом круге кровообращения.
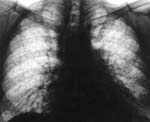
Важнейшими в диагностике являются рентгенологические методы обследования грудной клетки, особенно компьютерная томография высокого разрешения. На ранних стадиях болезни определяются преимущественно усиление и деформация легочного рисунка, понижение прозрачности легочных полей по типу «матового стекла», мелкоочаговые тени. По мере прогрессирования процесса деформация легочного рисунка становится более выраженной, выявляются признаки интерстициального фиброза, полостные образования, формируется картина «сотового легкого». Наиболее точная диагностика возможна при помощи оценки биопсийного материала. Биопсию легких долгое время считали «золотым стандартом» в диагностике идиопатического легочного фиброза, позволяющим не только установить диагноз, но и определить прогноз заболевания. Однако в последнее время эти позиции пересматриваются.
В качестве иллюстрации приводим историю болезни Сергея Е., 8 мес, находившегося на лечении в отделении реанимации Областной детской клинической больницы г. Курска. Ребенок от второй беременности, протекавшей на фоне тяжелой преэклампсии, от первых экстренных оперативных родов на сроке 36 нед, родился с массой 1520 г, длиной 43 см, оценка по шкале Апгар 1/4/7 баллов. Находился в отделении реанимации и интенсивной терапии, а в последующем – в отделении патологии недоношенных областного перинатального цента Курска в течение 1 мес; проводились респираторная поддержка искусственной вентиляций легких, антибактериальная, симптоматическая терапия. После выписки мальчик амбулаторно наблюдался педиатром по месту жительства, неоднократно находился на стационарном лечении с диагнозом рецидивирующий бронхит.
В возрасте 7 мес состояние ребенка ухудшилось – на фоне респираторной инфекции появился сухой частый кашель, в легких выслушивались сухие и влажные хрипы, в течение нескольких дней сохранялось повышение температуры тела до фебрильной. Отмечались вялость, общая слабость, снижение аппетита. Мальчик был госпитализирован в инфекционную больницу им. Н.А. Семашко с диагнозом: внебольничная правосторонняя среднедолевая пневмония. За время пребывания в состоянии ребенка на фоне терапии отмечалась отрицательная динамика – прогрессировали проявления дыхательной недостаточности, выраженного интоксикационного синдрома, и мальчик был переведен в областную детскую клиническую больницу, в отделение реанимации и интенсивной терапии.
Общее состояние ребенка при поступлении крайне тяжелое за счет проявлений тяжелой дыхательной и сердечной недостаточности с перегрузкой малого круга кровообращения. При осмотре физическое развитие дисгармоничное за счет дефицита массы тела 2-й степени. Масса 5840 г, рост 67 см (SDS массы –3,52; SDS роста –1,22). Кожные покровы и видимые слизистые оболочки бледные с мраморным рисунком, периоральный и периорбитальный цианоз, акроцианоз. Большой родничок 4,0×4,0 см, напряжен. Зев рыхлый, умеренная гиперемия дужек. Носовое дыхание затруднено, скудное слизистое отделяемое. Одышка в покое, усиливающаяся при физическом и психоэмоциональном напряжении. При кормлении быстро устает, вскармливается через зонд, объем питания – удерживает по 150 мл смеси через 3,5 ч, не срыгивает. Грудная клетка цилиндрическая. Дыхание с выраженным участием вспомогательной мускулатуры: втяжение межреберий, западение яремной ямки.
Перкуторно над легкими легочный звук с коробочным оттенком, местами с участками притупления. При аускультации дыхание жесткое, в нижнебоковых отделах ослаблено, выслушиваются крепитирующие хрипы с обеих сторон по всем полям. Ребенок получал респираторную поддержку, увлажненный кислород через носовые канюли со скоростью 3–3,5 л/мин. При этом сатурация крови кислородом (SpO2) 80–88%. При прекращении респираторной поддержки SpO2 снижался до 68–70%. Тоны сердца приглушены, ритм правильный, частота сердечных сокращений 138–180 в минуту, артериальное давление 101/66 мм рт.ст. Язык влажный. Живот мягкий, не вздут, безболезненный. Печень +2,5 см от края реберной дуги, селезенка не пальпируется. Стул был самостоятельно 2 раза, желтого цвета, водянистый, с примесью небольшого количества слизи, Мочеиспускание не нарушено. Диурез 2,4 мл/кг/ч.
При лабораторном обследовании в крови отмечались увеличение СОЭ до 63 мм/ч, лейкоцитоз до 13,5·109/л, проявления выраженного метаболического ацидоза, повышение активности трансаминаз, лактатдегидрогеназы, С-реактивного белка. Прокальцитониновый тест ≥2. При бактериологическом исследовании мокроты выделена Candida albicans, в отделяемом верхних дыхательных путей определялись Serrata rubidea, Stenotrophomonas maltophilia.
На рентгенограмме органов грудной клетки в прямой проекции отмечались инфильтративные изменения в средней доле правого легкого, субтотальное снижение пневматизации левого легкого. В дальнейшем отмечалась отрицательная динамика в виде нарастания отечно-инфильтративных изменений в легочной ткани с обеих сторон. На фоне тотального затемнения определялась воздушная бронхограмма. Тень средостения не дифференцировалась. Куполы диафрагмы не прослеживались.
По данным электрокардиографии определялись признаки увеличения правого желудочка; нарушение реполяризации. При ультразвуковом исследовании выявлены агенезия левой почки, компенсаторная гипертрофия правой почки, умеренная гепатомегалия, относительная недостаточность трикуспидального клапана. На компьютерной томограмме грудной клетки отмечены участки интерстициальных изменений, выраженные диффузные фиброзные изменения легочной ткани.
Ребенок был заочно проконсультирован в отделе хронических воспалительных и аллергических болезней легких НИКИ педиатрии им. акад. Ю.Е. Вельтищева. Был установлен клинический диагноз: идиопатический легочный фиброз (синдром Хаммана–Рича).
Данное редкое клиническое наблюдение подтверждает, что идиопатический легочный фиброз в раннем возрасте характеризуется быстро прогрессирующим течением, и прогноз остается неблагоприятным даже при своевременной верификации диагноза.
Миненкова Т.А., Мизерницкий Ю.Л., Разинькова Н.С., Сережкина А.В., Костюченко М.В.
Российский вестник перинатологии и педиатрии, 2019; 64:(4)
Что такое болезнь хаммена рича
Клиническая картина, сходная с диссеминированным туберкулезом определяется при прогрессирующем диффузном первичном интерстициалъном фиброзе легких, т. е. синдроме Хаммена — Рича. Это заболевание, этиология которого остается невыясненной, относят к аллергическим болезням или коллагенозам. При нем развивается сначала межальвеолярный отек, затем выпадает фибрин и происходит интенсивная пролиферация фибробластов, а также развивается соединительная ткань, что приводит к диффузному легочному фиброзу. В стенках альвеол наблюдается, кроме того, избыточное развитие эластина и коллагеновой субстанции, в них скопляется небольшое количество выпота с примесью макрофагов.
В результате организации выпота и фибрина в легких формируются множественные очаги и интерстициальные уплотнения в виде сетчатого склероза. Одновременно образуются мелкие кисты и участки буллезной эмфиземы, что придает легким характер «резиновой губки» или «медовых сот». Все эти морфологические изменения в своем рентгенологическом отображении могут походить на диссеминированный туберкулез с преобладанием фиброза.
Болезнь Хаммена — Рича встречается у лиц различного возраста, в том числе у детей и подростков. Но чаще она наблюдается в 30—50 лет, преимущественно у мужчин. У молодых она протекает большей частью остро с тенденцией к быстрому прогрессированию, у пожилых — скорее иодостро или хронически. Острое начало болезни характеризуется повышением температуры, надсадным сухим кашлем, резкой одышкой, цианозом, болями в груди, тахиспстолией и катаральными явлениями в легких в виде крепитации, напоминающей «треск целлофана». Эта клиническая картина па первых порах напоминает тяжелую форму пневмонии.
Однако антибактериальная терапия в таких случаях не только не оказывает эффекта, по, наоборот, нередко ухудшает состояние, возможно, в результате сенсибилизации организма больных. Лечение кортикосгероидными гормонами дает иногда временное и частичное улучшение.
При подостром и хроническом течении болезни развивается общая слабость, иногда появляется кровохарканье, нарастают тахикардия, цианоз н особенно одышка, причем не только при движении и физическом напряжении, но и в покое. Дыхание становится поверхностным, частота его достигает 40—50 в минуту, особенно затруднен глубокий вдох. Эти функциональные нарушения связаны с потерей эластичности легочной ткани, альвеолярно-капиллярным блоком, а на этой почве с нарушением диффузии газов, артериальной и тканевой гипоксией. Довольно быстро утолщаются концевые фаланги пальцев рук и ног по типу «барабанных палочек».
В гемограмме определяются умеренный лейкоцитоз и полицитемия, СОЭ ускорена (25—50 мм/ч). В сыворотке крови повышено содержание у-глобулинов (до 25—30%) и фибриногена. По мере эволюции болезни, которая при острой форме длится около 6 мес, при подострой до 2 лет, а при хронической — 2—3—5 лет и более, развивается правожелудочковая недостаточность, что в конце концов приводит к летальному исходу.
Хотя некоторые из указанных симптомов наблюдаются и при подостром пли хроническом диссеминированном туберкулезе легких, ряд признаков отличают его от болезни Хаммена — Рича, при которой отмечаются тяжелая одышка и резко выраженный цианоз, часто отрицательные туберкулиновые пробы, высокая гиперглобулинемия, отсутствие микобактерий туберкулеза в мокроте и неэффективность специфической химиотерапии. Своеобразна при этой болезни рентгенологическая картина.
За диссеминированный туберкулез иногда принимают изменения в легких, возникающее при различных больших коллагенозах. Поводом для этого служат также некоторые общие клинические симптомы: лихорадка, кашель, иногда кровохарканье, боли в груди, похудание, катаральные явления в легких и др. Предположение о туберкулезе укрепляется при обнаружении на рентгенотрамме расеянных очаговых изменений, участков уплотнения или распада легочной ткани, плеврита, спонтанного пневмоторакса. Трудности распознавания природы процесса усугубляются при сочетании коллагеновой болезни и туберкулеза. При их дифференциальной диагностике следует учитывать ряд признаков, в большей мере характерных для коллагенозов, чем для туберкулеза.
К ним относятся лихорадка, часто неправильного типа, сопровождающаяся ознобом; аллергические и аутоиммунные реакции в виде кожных сыпей, артралгий и мигрирующих артритов; отсутствие эффекта от антибиотиков и, наоборот, положительное действие противовоспалительных и особенно глюкокортикоидных препаратов; снижение чувствительности к туберкулину; высокое содержание в крови фибриногена, альфа- и особенно у-глобулинов, С-реактивного белка. Помимо этих общих симптомов, следует учитывать клинические особенности, присущие отдельным видам коллагенозов. Для ревматизма, например, согласно схеме Джонса, несколько измененной А. И. Нестеровым и Я. А. Сигидиным (1966), характерны кардит, полиартрит, хорея, аннулярная эритема, высокие показатели антистрептолизииа О и антигиалуронидазы и др.
Все эти признаки отсутствуют при туберкулезе. Отличаются от последнего и рентгенологические проявления ревматизма легкого, который встречается у 20—50% больных. Ревматическая пневмония, или пневмониты, возникающие обычно при первых или повторных атаках болезни, имеют характер диффузно-интерстициального уплотнения преимущественно в нижних отделах легких. Здесь же локализуются очаговоподобные изменения за счет множественных васкулитов. Возникающий плеврит обычно сочетается с другими проявлениями ревматизма и часто бывает двусторонним. Выпот в плевральных полостях небольшой и быстро рассасывается, не оставляя после себя массивных плевральных сращений. Все эти симптомы не характерны для туберкулеза.
Фиброзирующий альвеолит – это патологический процесс, в основе которого лежит обширное поражение интерстициальной ткани легких, приводящее к развитию фиброзных изменений и дыхательной недостаточности. Прогрессирование фиброзирующего альвеолита сопровождается неуклонным нарастанием слабости, похудания, одышки, малопродуктивного кашля, болей в грудной клетке, цианоза. Диагностика основывается на данных рентгенографии и компьютерной томографии легких, спирометрии, биопсии легких. Лечение фиброзирующего альвеолита включает противовоспалительную и иммуносупрессивную терапию, оксигенотерапию; по показаниям – трансплантацию легких.
МКБ-10
Общие сведения
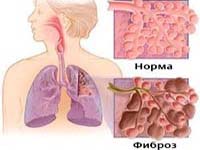
Альвеолит фиброзирующий идиопатический (синонимы: фиброз легочный идиопатический, синдром Хаммена-Рича) – прогрессирующее диффузное двухстороннее поражение альвеол и интерстициальной легочной ткани, сопровождающееся развитием диффузного фиброза и нарастающей дыхательной недостаточности. Фиброзирующий альвеолит имеет исключительно легочную локализацию, плохо поддается терапии, часто оканчивается летальным исходом.
Фиброзирующий альвеолит – относительно редкое заболевание неясной этиологии, однако имеет тенденцию к возрастанию. Фиброзирующий альвеолит чаще поражает мужчин старше 50 лет (20 случаев из 100 тыс.), чем женщин (13 случаев из 100 тыс.). Летальность при фиброзирующем альвеолите достигает 3,3 случая на 100 тыс. населения.
Причины
Причины возникновения фиброзирующего альвеолита неясны. Существуют предположения об аутоиммунном характере заболевания, вирусной природе (герпесвирус, вирус гепатита С, аденовирусы, цитомегаловирус), наследственной предрасположенности.
В распространённости фиброзирующего альвеолита имеют значение профессиональные, экологические, бытовые и географические факторы. Так, замечено, что развитию идиопатического фиброзирующего альвеолита в наибольшей степени подвержены фермеры, разводящие птиц, рабочие, контактирующие с древесной, асбестовой, металлической и силикатной пылью, курящие пациенты.
Патогенез
Воспалительные явления в альвеолах вызывают необратимое утолщение их стенок и снижение проницаемости для газообмена. Развитие идиопатического фиброзирующего альвеолита характеризуется тремя взаимосвязанными процессами: интерстициальным отеком, интерстициальным воспалением (альвеолитом) и интерстициальным фиброзом.
В острой стадии интерстициального отека происходит поражение альвеолярных капилляров и эпителия, их отек и формирование гиалиново-мембранных комплексов, препятствующих расширению альвеолярной ткани при дыхании. На этой стадии возможен регресс процесса либо развитие интерстициальной пневмонии. Хроническая стадия интерстициального воспаления характеризуется дальнейшим прогрессированием процесса, отложением в альвеолах коллагена и развитием распространенного фиброза с обширным повреждение легочной ткани.
В терминальной стадии интерстициального фиброза происходит полное замещение капиллярной сети и альвеолярной ткани фиброзной с формированием полостных расширений. Ткань легкого напоминает по внешнему виду пчелиные соты. Необратимые изменения в альвеолярно-капилярной системе легких при фиброзирующем альвеолите приводят к рестриктивным изменениям, расстройству газообмена, прогрессированию дыхательной недостаточности и к гибели пациента.
Классификация
В клинической пульмонологии выделяют 3 формы фиброзирующих альвеолитов:
Симптомы фиброзирующего альвеолита
Развитие заболевания постепенное с развитием необратимых изменений в альвеолах и неуклонным нарастанием одышки. Длительное время пациенты не придают этому значения и не обращаются к врачу, объясняя изменения в своем самочувствии усталостью на работе, прекращением спортивных занятий, набором веса и т. д. Обычно от начала первых симптомов фиброзирующего альвеолита до обращения в медицинское учреждение проходит от 3 месяцев до 1-3 лет.
Ведущими жалобами при фиброзирующем альвеолите служат выраженная одышка при минимальных физических нагрузках и малопродуктивный кашель. Пациентов беспокоят боли в грудной клетке (под лопатками), препятствующие глубокому вдоху, похудание, слабость, мышечные и суставные боли, повышенная температура тела. У половины пациентов с фиброзирующим альвеолитом ногтевые фаланги приобретают характерный вид «барабанных палочек» с «часовыми стеклами». Цианоз и одышка резистентны к проводимой терапии. В дальнейшем происходит нарастание гипоксемии, легочной гипертензии и правожелудочковой недостаточности.
Осложнения
В терминальной стадии фиброзирующего альвеолита усиливаются признаки дыхательной недостаточности и развития легочного сердца: серо-пепельный диффузный цианоз, набухание вен шеи, отеки, развитие кахексии. Течение фиброзирующего альвеолита вызывает прогрессирующую дыхательную недостаточность, развитие легочного сердца, может осложниться отеком легких.
Диагностика
При фиброзирующем альвеолите в легких выслушивается жесткое дыхание, крепитация (симптом «треска целлофана») и сухие хрипы; перкуторный звук укорочен. Изменения со стороны сердца характеризуются тахикардией и приглушенностью тонов. Этапы диагностики:
Проведение открытой биопсии легких при фиброзирующем альвеолите позволяет определить стадию болезни, выбор соответствующего лечения, развитие заболевания. При гистологическом исследовании выделяют пять степеней изменений:
Дифференциальная диагностика
Фиброзирующий альвеолит следует дифференцировать с пневмонией, гранулематозом, саркоидозом, диссеминированным туберкулезом, бронхиолоальвеолярным раком, пневмокониозом, диффузным амилоидозом и др.
Выработаны большие и малые критерии диагностики фиброзирующего альвеолита. С наибольшей вероятностью фиброзирующий альвеолит может быть диагностирован при определении 4-х больших и 3-х малых критериев. Большими критериями верификации идеопатического фиброзирующего альвеолита служат:
К малым критериям идеопатического фиброзирующего альвеолита относятся:
Лечение фиброзирующего альвеолита
Немедикаментозная терапия
Целью проводимого врачом-пульмонологом лечения является облегчение течения заболевания, замедление прогрессирования фиброза легочной ткани, качественное улучшение жизни пациента. К немедикаментозному лечению фиброзирующего альвеолита относятся специальные реабилитационные программы, включающие физические тренировки, психологическую поддержку пациентов. Выраженным терапевтическим эффектом обладает кислородная терапия, уменьшающая одышку и повышающая выносливость физических нагрузок у пациентов с фиброзирующим альвеолитом.
Фармакотерапия
Для достижения состояния ремиссии в медикаментозную терапию фиброзирующего альвеолита включают противовоспалительные (глюкокортикостероиды) и антифиброзные (пеницилламин, колхицин) препараты, иммунодепрессанты (азатиоприн), а также их комбинацию. Одновременно назначаются верошпирон, препараты калия, пиридоксин, бронхолитики. Медикаментозная терапия дает эффект лишь в случаях отсутствия выраженного пневмофиброза. С целью предупреждения вирусных инфекций всем пациентам с фиброзирующим альвеолитом показана противогриппозная и антипневмококковая вакцинация.
Радикальное лечение фиброзирующего альвеолита предусматривает трансплантацию легких, дающую высокий процент 5-летней выживаемости – до 50-60%. Показаниями к пересадке легких служат выраженные гипоксемия, диспноэ, снижение ЖЕЛ ниже 70%, снижение диффузной способности легких.
Прогноз и профилактика
В течении идиопатического фиброзирующего альвеолита после проведения лекарственной терапии наблюдаются периоды ремиссии, однако заболевание все равно постепенно прогрессирует. Средний процент выживаемости при диагностике нелеченных фиброзирующих альвеолитов составляет 3-4 года. Более благоприятным течением отличаются аллергические и токсические альвеолиты, которые при устранении провоцирующего фактора на ранних стадиях могут регрессировать.
Меры профилактики идиопатического фиброзирующего альвеолита включают предупреждение инфекций, исключении вредных профессиональных, экологических и бытовых факторов. Пациенты с фиброзирующим альвеолитом должны находиться на диспансерном учете у пульмонолога, аллерголога и врача-профпатолога.



