Паренхиматозная белковая дистрофия
Паренхиматозная белковая дистрофия проявляется появлением в цитоплазме большого количества зёрен белковой природы. Возникает в печени, почках, сердце, при инфекции и интоксикации. Дистрофия обратимая после прекращения действия травмируемого фактора.
Содержание
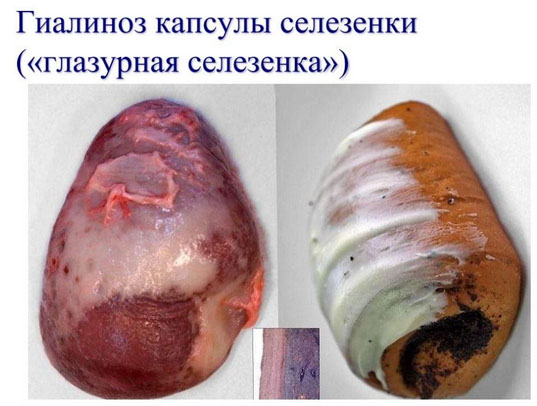
Гиалиново-капельная
Проявляется появлением в клетках капель, напоминающие гиалиновый хрящ. Возникает в почках, печени при хронических заболеваниях этих органов. Дистрофия — необратимая.
Зернистая дистрофия
Проявляется появлением в цитоплазме большого количества зёрен белковой природы. Возникает в печени, почках, сердце, при инфекции и интоксикации. Дистрофия обратимая после прекращения действия травмируемого фактора.
Примечания
Ссылки

Полезное
Смотреть что такое «Паренхиматозная белковая дистрофия» в других словарях:
Дистрофия — (др. греч. dystrophe, от dys… приставка, означающая затруднение, нарушение, и trophe питание) патологический процесс, в результате которого та или иная ткань теряет или накапливает вещества, в норме не характерные для неё… … Википедия
Гиперпротеинемия — (новолат. hyperproteinaemia) повышенная концентрация белков в крови. Гиперпротеинемия возникает на фоне других патологических процессов, таких как гемобластоз, миеломная болезнь, болезнь Вальденстрема, сгущении крови.[1] Белковая… … Википедия
Паренхиматозные дистрофии
В зависимости от нарушений того или иного вида обмена паренхиматозные дистрофии делят на белковые (диспротеинозы), жировые (липидозы) и углеводные.
Паренхиматозные белковые дистрофии (диспротеинозы)
Большая часть белков цитоплазмы (простых и сложных) находится в соединении с липидами, образуя липопротеидные комплексы. Эти комплексы составляют основу мембран митохондрий, эндоплазматическои сети, пластинчатого комплекса и других структур. Помимо связанных белков, в цитоплазме содержатся и свободные. Многие из последних обладают функцией ферментов.
Сущность паренхиматозных диспротеинозов состоит в изменении физико-химических и морфологических свойств белков клетки: они подвергаются
К паренхиматозным диспротеинозам относят гиалиново-капельную, гидропическую и роговую дистрофии.
К паренхиматозным белковым дистрофиям со времен Р. Вирхова причисляли и многие патологи продолжают причислять так называемую зернистую дистрофию, при которой в клетках паренхиматозных органов появляются белковые зерна. Сами органы увеличиваются в размерах, становятся дряблыми и тусклыми на разрезе, что послужило причиной называть также зернистую дистрофию тусклым (мутным) набуханием. Однако электронно-микроскопическое и гистоферменто-химическое изучение «зернистой дистрофии» показало, что в ее основе лежит не накопление белка в цитоплазме, а гиперплазия ультраструктур клеток паренхиматозных органов как выражение функционального напряжения этих органов в ответ на различные воздействия; гиперплазированные ультраструктуры клетки выявляются при светооптическом исследовании как белковые гранулы.
При гиалиново-капельной дистрофии в цитоплазме появляются крупные гиалиноподобные белковые капли, сливающиеся между собой и заполняющие тело клетки; при этом происходит деструкция ультраструктурных элементов клетки. В ряде случаев гиалиново-капельная дистрофия завершается фокальным коагуляционным некрозом клетки.
В почках при микроскопическом исследовании накопление гиалиновых капель находят в нефроцитах. При этом наблюдается деструкция митохондрий, эндоплазматической сети, щеточной каемки. В основе гиалиново-капельной дистрофии нефроцитов лежит недостаточность вакуолярно-лизосомального аппарата эпителия проксимальных канальцев, в норме реабсорбирующего белки. Поэтому этот вид дистрофии нефроцитов очень часто встречается при нефротическом синдроме. Этот синдром является одним из проявлений многих заболеваний почек, при которых первично поражается гломерулярный фильтр (гломерулонефрит, амилоидоз почек, парапротеинемическая нефропатия и др.).
Внешний вид почек при этой дистрофии не имеет каких-либо характерных черт, он определяется прежде всего особенностями основного заболевания (гломерулонефрит, амилоидоз).
Внешний вид печени различен; изменения характерны для тех ее заболеваний, при которых встречается гиалиново-капельная дистрофия.
Исход гиалиново-капельной дистрофии неблагоприятен: она завершается необратимым процессом, ведущим к некрозу клетки.
Функциональное значение этой дистрофии очень велико. С гиалиново-капельной дистрофией эпителия почечных канальцев связаны появление в моче белка (протеинурия) и цилиндров (цилиндрурия), потеря белков плазмы (гипопротеинемия), нарушение ее электролитного баланса. Гиалиново-капельная дистрофия гепатоцитов нередко является морфологической основой нарушений многих функций печени.
Гидропическая, или водяночная, дистрофия характеризуется появлением в клетке вакуолей, наполненных цитоплазматической жидкостью. Она наблюдается чаще в эпителии кожи и почечных канальцев, в гепатоцитах, мышечных и нервных клетках, а также в клетках коры надпочечников.
Микроскопическая картина: паренхиматозные клетки увеличены в объеме, цитоплазма их заполнена вакуолями, содержащими прозрачную жидкость. Ядро смещается на периферию, иногда вакуолизируется или сморщивается. Прогрессирование этих изменений приводит к распаду ультраструктур клетки и переполнению клетки водой. Клетка превращается в заполненные жидкостью баллоны или в огромную вакуоль, в которой плавает пузырьковидное ядро. Такие изменения клетки, которые по существу являются выражением фокального колликвационного некроза, называют баллонной дистрофией.
Внешний вид органов и тканей мало изменяется при гидропической дистрофии, она обнаруживается обычно под микроскопом.
Механизм развития гидропической дистрофии сложен и отражает нарушения водно-электролитного и белкового обмена, ведущие к изменению коллоидно-осмотического давления в клетке. Большую роль играет нарушение проницаемости мембран клетки, сопровождающееся их распадом. Это ведет к закислению цитоплазмы, активации гидролитических ферментов лизосом, которые разрывают внутримолекулярные связи с присоединением воды.
Исход гидропической дистрофии, как правило, неблагоприятный; она завершается фокальным или тотальным некрозом клетки. Поэтому функция органов и тканей при гидропической дистрофии резко страдает.
Роговая дистрофия, или патологическое ороговение, характеризуется избыточным образованием рогового вещества в ороговевающем эпителии (гиперкератоз, ихтиоз) или образованием рогового вещества там, где в норме его не бывает (патологическое ороговение на слизистых оболочках, или лейкоплакия; образование «раковых жемчужин» в плоскоклеточном раке). Процесс может быть местным или распространенным.
Причины роговой дистрофии разнообразны: нарушение развития кожи, хроническое воспаление, вирусные инфекции, авитаминозы и др.
Исход может быть двояким: устранение вызывающей причины в начале процесса может привести к восстановлению ткани, однако в далеко зашедших случаях наступает гибель клеток.
Значение роговой дистрофии определяется ее степенью, распространенностью и длительностью. Длительно существующее патологическое ороговение слизистой оболочки (лейкоплакия) может явиться источником развития раковой опухоли. Врожденный ихтиоз резкой степени, как правило, несовместим с жизнью.
К группе паренхиматозных диспротеинозов примыкает ряд дистрофий, в основе которых лежат нарушения внутриклеточного метаболизма ряда аминокислот в результате наследственной недостаточности метаболизирующих их ферментов, т. е. в результате наследственной ферментопатии. Эти дистрофии относятся к так называемым болезням накопления.
Наиболее яркими примерами наследственных дистрофий, связанных с нарушением внутриклеточного метаболизма аминокислот, являются цистиноз, тирозиноз, фенилпировиноградная олигофрения (фенилкетонурия).
Наследственные дистрофии, связанные с нарушением обмена аминокислот
Дистрофия

Общие сведения
Что такое дистрофия? Дистрофия (лат. Dystrophia) представляет собой патологический процесс, обусловленный количественными/качественными изменениями в метаболизме клеток тканей/межклеточном веществе, приводящие к их структурным изменениям. В обиходе часто можно встретить выражение «дистрофик» или ты можешь стать дистрофиком применительно к очень худому человеку, то есть, это мужчины или женщины с крайней степенью истощения организма. Непосредственной причиной дистрофий являются различного рода нарушения клеточных/внеклеточных механизмов, обеспечивающих трофику тканей. Изучением дистрофических процессов занимается такая область медицинских знаний, как патологическая анатомия человека, изучающая морфологические/структурные изменения в тканях организма, возникающие в результате воздействия экзогенных/эндогенных факторов и их влияние на развитие болезнетворного процесса.
В основе дистрофии могут лежать различные внутриклеточные/внеклеточные процессы аккумуляции/уменьшения различных веществ: жидкости, белков, жиров, липидов, углеводов; пигментов; образование аномальных веществ (например, продуктов нарушенного метаболизма); процессы распада различного рода структур. Дистрофические процессы могут протекать в тканях различных органов, формируя ту или иную патологию, например:
При этом, они могут протекать преимущественно в паренхиме, строме или одновременно в паренхиме и строме, в соответствии с чем выделяют определенные виды дистрофий.
Паренхиматозные дистрофии
Для этого вида дистрофий характерно нарушения обмена в паренхиматозных органах (почки, сердце, печень). В основе паренхиматозных дистрофий лежит недостаточность конкретного ферментативного механизма (ферментопатии приобретенного/наследственного генеза), необходимого для выполнения специфических функций клеток (кардиомиоцитов гепатоцитов, нефроцитов). Паренхиматозные дистрофии включают:
паренхиматозные диспротеинозы (син. белковая дистрофия), сопровождающиеся появлением включений белковой природы в цитоплазму клеток, гидратацией клетки в них накоплением ионов натрия. Морфологически это гиалиново-капельная дистрофия, гидропическая дистрофия, роговая и зернистая дистрофия.
Гиалиново-капельная дистрофия характеризуется появлением в цитоплазме клетки гиалиноподобных крупных включений белка и всегда приводит к смерти клетки в то время, как при гидропической дистрофии в клеточной цитоплазме появляются вакуоли (син. вакуольная дистрофия), которая зачастую завершается развитием баллонной дистрофии с последующей смертью клетки в результате колликвационного некроза. Развивается в почках при нефротическом синдроме, гломерулонефрите, амилоидозе (дистрофия эпителия канальцев); в печени при вирусном, алкогольном гепатите, холестазе, билиарном циррозе (дистрофия в гепатоцитах) и значительно реже встречаются в сердечной мышце.
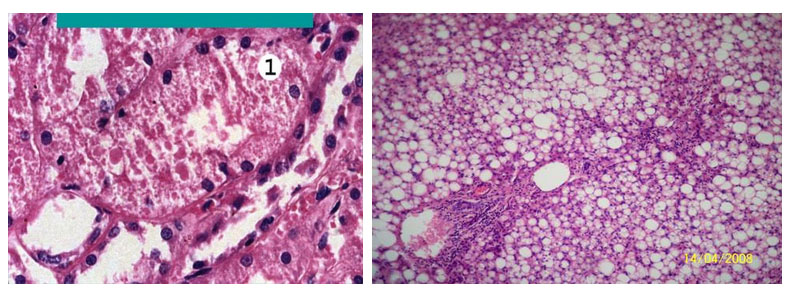
Гиалиново-капельная дистрофия и гидропическая (вакуольная) дистрофия
Зернистая дистрофия, наоборот, относится к поверхностной и в большинстве случаев при устранении причины является обратимой дистрофией, однако, при продолжающемся патологическом действии процесс может стать необратимым, преобразуясь в некроз. Органы при ней становятся немного набухшими, а при разрезе поверхность выглядит мутной и тусклой. Ее развитию способствуют различного рода инфекции и интоксикации. Развивается чаще в почках (зернистая дистрофия эпителия извитых канальцев почки), реже в печени, вызывая дискомплексацию печеночных балок и в сердечной мышце и этом, дистрофия сердечной мышцы сильно не выражена (фото ниже).
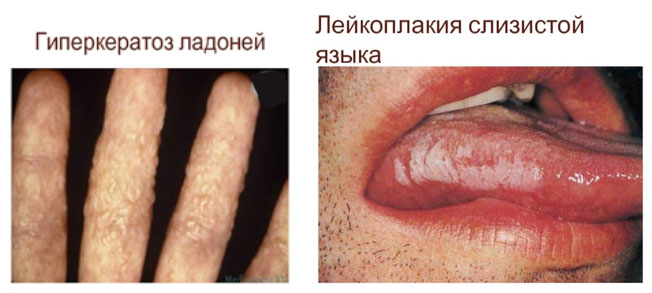
 Роговая дистрофия (син. патологическое ороговение). Для нее характерно избыточная выработка в ороговевающем эпителии рогового вещества, что проявляется такими заболеваниями как (ихтиоз/гиперкератоз) или его там, где его в норме не должно быть (лейкоплакия, патологическое ороговение слизистых — гиперкератоз). При этом, патологический может быть распространенным или локализованным. Основными причинами развития роговой дистрофии являются вирусные инфекции, хроническое воспаление, нарушение развития кожи, авитаминозы. Исходом этого вида дистрофии в начале процесса может быть восстановление ткани при устранении причины, а в запущенных случаях клети погибают (фото ниже).
Роговая дистрофия (син. патологическое ороговение). Для нее характерно избыточная выработка в ороговевающем эпителии рогового вещества, что проявляется такими заболеваниями как (ихтиоз/гиперкератоз) или его там, где его в норме не должно быть (лейкоплакия, патологическое ороговение слизистых — гиперкератоз). При этом, патологический может быть распространенным или локализованным. Основными причинами развития роговой дистрофии являются вирусные инфекции, хроническое воспаление, нарушение развития кожи, авитаминозы. Исходом этого вида дистрофии в начале процесса может быть восстановление ткани при устранении причины, а в запущенных случаях клети погибают (фото ниже).
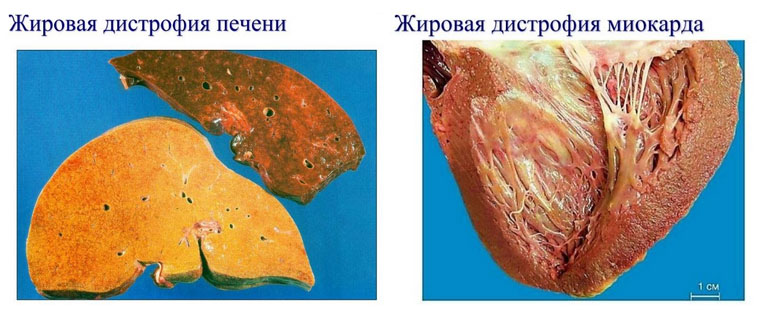
 Паренхиматозные липидозы (син. жировая дистрофия). Для них характерно нарушения жирового обмена в цитоплазме вследствие дисбаланса между процессами поступления/утилизации и выведением липидов в гепатоцитах, что проявляется накоплением капель триглицеридов в цитоплазме клеток. Жировая дистрофия развивается чаще в печени, почках, миокарде и проявляется жировой дистрофией печени при общем ожирении, сахарном диабете, алкоголизме, анемии, гипоксии, недостатке в пище белка, заболеваниях ЖКТ, действии токсичных веществ (фосфор, этанол, углерод). Жировая дистрофия печени в большинстве случаев обратима, однако при присоединении некроза, возможно, развития печеночной недостаточности. Жировая дистрофия миокарда развивается преимущественно на фоне гипоксии (ХСН, анемии), интоксикаций (отравление мышьяком/фосфором, дифтерийная, алкогольная дистрофия).
Паренхиматозные липидозы (син. жировая дистрофия). Для них характерно нарушения жирового обмена в цитоплазме вследствие дисбаланса между процессами поступления/утилизации и выведением липидов в гепатоцитах, что проявляется накоплением капель триглицеридов в цитоплазме клеток. Жировая дистрофия развивается чаще в печени, почках, миокарде и проявляется жировой дистрофией печени при общем ожирении, сахарном диабете, алкоголизме, анемии, гипоксии, недостатке в пище белка, заболеваниях ЖКТ, действии токсичных веществ (фосфор, этанол, углерод). Жировая дистрофия печени в большинстве случаев обратима, однако при присоединении некроза, возможно, развития печеночной недостаточности. Жировая дистрофия миокарда развивается преимущественно на фоне гипоксии (ХСН, анемии), интоксикаций (отравление мышьяком/фосфором, дифтерийная, алкогольная дистрофия).
Углеводные дистрофии. Для этого вида дистрофий характерно нарушением обмена гликогена/гликопротеидов. К углеводным паренхиматозным дистрофиям, обусловленных нарушением процесса обмена гликогена относятся сахарный диабет и гликогенозы (наследственные углеводные дистрофии).
Развивающаяся абсолютная/относительная недостаточность инсулина при сахарном диабете вызывает нарушение утилизация глюкозы и процесса синтез гликогена, что приводит к повышается в крови содержания глюкозы и ее появлению в моче на фоне истощения запасов гликогена в мышцах/печени. Поражаются почечные канальцы, а также клубочки, у которых базальная мембрана становится более проницаемой для белков плазмы/сахаров. Нарушение процесса синтеза гликогена в печени способствует ее инфильтрации жирами с постепенным развитием жировой дистрофии печени.
Гликогенозы развиваются при наследственных заболеваниях (болезни накопления, ферментопатии) и обусловлены нарушениями обмена гликогена. Избыточное накопление гликогена отмечается преимущественно в скелетных мышцах, клетках миокарда, печени/почках, что вызывает вторичное повреждение клетки и некроз.
Паренхиматозные углеводные дистрофии, обусловленные нарушением обмена гликопротеидов, проявляются накоплением в клетках мукоидов/муцинов (слизистых веществ), поэтому зачастую ее называют слизистой дистрофией. Усиленное образование/изменение физико-химических свойств слизи приводит к гибели клеток.
Примером может быть слизистая дистрофия при муковисцидозе, характеризующегося изменением качества слизи (высокая вязкость, густая, тяжело выводится), что способствует развитию ретенционных кист/склероза (кистозный фиброз).
Стромально-сосудистые дистрофии (син. мезенхимальные дистрофии). Дистрофии такого рода обусловлены нарушениями обмена веществ непосредственно в интерстициальной соединительной ткани, состоящей из эластина, белков коллагена и гликозаминогликанов, формирующих строму органов/стенок сосудов. Для этой группе дистрофий характерно накопление в соединительной ткани различных продуктов метаболизма, что способствует развитию патологических состояний. Стромально-сосудистые дистрофии также подразделяют на белковые, жировые и углеводные.
Стромально-сосудистые диспротеинозы. В группе диспротеинозов выделяют:
Мезенхимальные липидозы
Жировые мезенхимальные дистрофии развиваются на фоне нарушений обмена нейтрального жира/холестерина. Нейтральный (запасный жир) в норме накапливается в жировых депо для обеспечения энергетических потребностей человеческого организма. В основе дистрофии этого типа избыточное накопление жира в тканях, где он в норме должен отсутствовать или уменьшение его количества. Общее увеличение нейтрального жира проявляется тучностью/общим ожирением. Основными причинами являются нейроэндокринные нарушения процесса регуляции жирового обмена, возникающие на фоне поражения гипофиза, заболеваний ЦНС, избыточного питания (алиментарное ожирение). К тяжёлым правлениям относится ожирение сердца, когда жир откладывается между мышечными волокнами/под эпикардом, что приводит к атрофии миокарда и резкому снижению его функции. Нарушение холестеринового обмена приводит к его очаговым накоплениям в интиме крупных кровеносных сосудов, что является основой развития атеросклероза (рис. ниже).
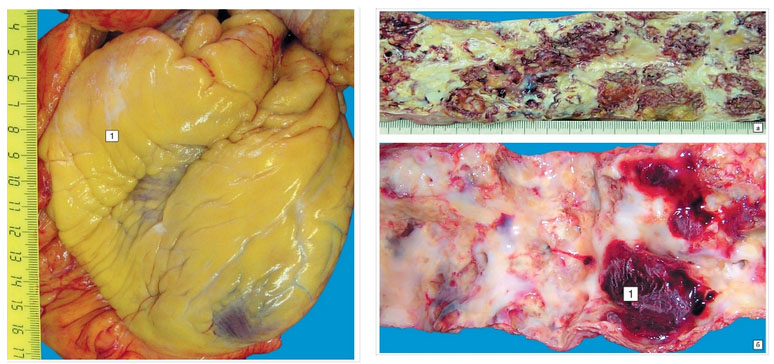
Ожирение сердца, атеросклероз аорты с липидными/атеросклеротическими изменениям
Мезенхимальные углеводные дистрофии. В их основе нарушение обмена гликопротеидов/гликозаминогликанов. Проявлением этого вида дистрофии является развитие густой слизеподобной массы на месте соединительной ткани и жировой клетчатки, что обусловлено высвобождением хромотропных веществ из связей с белками, которые накапливаются в межуточном веществе и постепенно происходит замещение коллагеновых волокон соединительной/жировая ткани, стромы органов на слизеподобную массу. Обусловлена нарушением эндокринных желёз (микседема, кахексия, болезни накопления).
Смешанные дистрофии
Смешанные дистрофии являются морфологическим проявлением расстройства метаболизма преимущественно сложных белков (нуклеопротеидов, эндогенных пигментов-хромопротеидов, а также липопротеидов и минералов как в паренхиме, происходящие так и в стенках сосудов/строме органов и тканей). Наиболее часто манифестируют различными нарушениями эндогенных пигментов (хромопротеидов), то есть, синтезируемых непосредственно в организме и придающих тканям/органам различную окраску (гемоглобин, ферритин, гемосидерин, гематоидин, гемин, порфирины и др.). На практике проявляются такими нарушениями как:
Существует и множество других видов расстройств (гипермеланоз, невис, меланома, липофусциноз и др.), однако они не являются темой этой статьи.
Патогенез
Среди патогенетических механизмов лежащих в основе дистрофических изменений выделяют:
Установлено, что основными морфогенетическими механизмами дистрофий являются инфильтрация, фанероз и зачастую они относятся к различным стадиям развития патологического процесса. Но в некоторых тканях/органах в силу их структурно-функциональной специфики преобладает один из вышеописанных морфогенетических механизмов.
Классификация
В основу классификации дистрофий положены различные признаки, на основании которых принято выделять различные виды дистрофии:
Причины
Дистрофии относятся к полиэтиологическим патологическим процессам. Каждый вид дистрофии имеет свои причины развития. При этом, причины, способные потенциально вызывать дистрофию, могут действовать непосредственно или через рефлекторные/гуморальные механизмы, то есть, опосредованно. Степень/характер повреждения определяется природой/длительностью действия и силой повреждающего фактора, особенностями структуры/функции ткани или органа и реактивностью организма. Соответственно, при некоторых видах дистрофий возникают обратимые поверхностные изменения, при других — необратимые глубокие, сопровождающиеся высоким риском гибели клеток/тканей, а зачастую и целых органов. Причины развития дистрофий чрезвычайно разнообразны, среди которых можно выделить основные:
Те или иные повреждающие факторы, воздействуя непосредственно на биохимические процессы межклеточных структур/клетки способствуют как морфологическим изменениям, так и функциональным нарушениям. При этом, точный момент, при котором дистрофические процессы становятся необратимыми остается до настоящего времени неизвестным.
Симптомы
Поскольку дистрофия, как патологический процесс является составляющим элементом патогенеза большого числа заболеваний рассмотреть их симптоматику в короткой статье не представляется возможным. В качестве примера, рассмотрим лишь болезнь Фридриха и миотоническую дистрофию.
Наследственная атаксия Фридрейха
Атаксия Фридрейха (АФ) относится к врожденным заболеваниям, передаваемых по аутосомно-рецессивному типу то есть, дети с этой патологией рождаются у пары клинически здоровых родителей, но которые являются носителями гена, вызывающего патологию. Наиболее часто встречаемая врожденная атаксия: показатель встречаемости варьирует в пределах около 2-7 случаев/100 тысяч человек. При АФ поражаются:
Протекает на фоне прогрессирующей дегенерации ЦНС и периферической нервной системы. Симптоматика заболевания появляются преимущественно на 1-2 десятилетии жизни, реже — позже. Основными признаками являются пошатывание/неуверенность, спотыкание и частые падения во время ходьбы, слабость в ногах, тремор, дизартрия, нарушения слуха. Постепенно исчезают надкостничные/сухожильные рефлексы (ахилловы/коленные). Зачастую ранним проявлением является ревмокардит, нистагм, расстройства сидения. При физикальном обследовании пациенты не могут выполнить пяточно-коленную пробу, отмечается выраженное покачивание в позе Ромберга, усиливающееся при закрывании глаз.
Постепенно нарастает нарушение глубокой чувствительности, прогрессирует мышечная атрофия, которая первоначально превалирует на нижних конечностях, а позже распространяется и на верхние конечности. Быстро формируется тотальная арефлексия, развивается катаракта и атрофируется зрительный нерв, что приводит к слепоте, деменции, нарушению функция тазовых органов. Практически параллельно развиваются эндокринные нарушения в виде дисфункции яичников, гипогонадизма, сахарного диабета. Появляются кардиомиопатии и различного рода деформации скелета: кифосколиоз, искривление позвоночника, «стопа Фридрейха», косолапость, деформация пальцев рук/ног. Болезнь неуклонно прогрессирует, в случаях отсутствии своевременного/адекватного лечения, длительность болезни не превышает 20 лет, реже пациенты доживают до 70-80 лет. Непосредственной причиной смерти являются инфекционные осложнения и сердечно-легочная недостаточность.
Миотоническая дистрофия
Клинические проявления МД манифестируют мышечной (миотония, миопатия, миалгия) и вне мышечной симптоматикой в виде с вегетативно-трофических нарушений. При этом, проявления заболевания несколько отличаются в зависимости от типа (1 и 2 тип). В основе заболевания мутации на длинном плече хромосом в различных генах. Тип 1 протекает более тяжело, чем тип 2.
Начало заболевания существенно варьирующим: от периода пренатальности до 50-60 лет. По возрасту начала заболевания выделяют 4 формы: врожденная (манифестирует сразу после рождения ребенка), юношеская (дебют заболевания в период от года до13-15 лет), классическая форма (20-30 лет) и минимальная форма, проявляющаяся в 50-60 лет. Кроме различия в возрасте манифестации проявления заболевания также имеются различия в симптоматике этого заболевания в зависимости от формы. К общим признакам заболевания относятся:
миотонический/миопатический синдромы. Характерно поражение мышц дистальных отделов верхних/нижних конечностей, лица, дыхательной мускулатуры, что выражается миотоническими спазмами в мышцах-сгибателях рук/ног, преимущественно в пальцах, а также жевательных мышцах, что проявляется проявляются неспособностью мышц к расслаблению (релаксации) после их сокращения при попытке открыть рот, разжать кулак, свистнуть.
На поздних стадиях заболевания проявления активной миотонии исчезают по мере нарастания мышечной слабости/атрофии. Появляется слабость и симметричные мышечные подергивания, атрофия выражены преимущественно в височных, жевательных, надостных, грудинно-сосцевидных мышцах, реже наблюдается атрофии круговой мышцы глаза, мимических мышц, вследствие чего лицо приобретает угрюмо-печальное маскообразное выражение. При поражении бульбарных мышц изменяется оттенок голоса, речь делается монотонной/малоразборчивой, возникает поперхивание, затруднение глотания. Миотонические спазмы дыхательных мышц и их слабость способствуют ограничению движений грудной клетки и нарушению вдоха/выдоха, что снижает легочную вентиляцию, изменяет частоту, ритм и амплитуду дыхания.
Из вне мышечной симптоматики характерно нарушение функции ЦНС, что проявляется сонливостью, постоянной усталостью, апатией. Часто выявляется снижение интеллекта/задержка психического развития. Наиболее частыми являются симптомы со стороны сердца (кардиомиопатия, аритмии, застойная сердечная недостаточность, атриовентрикулярные блокады). Из респираторной симптоматики — гиповентиляции, апноэ во время сна и аспирационная пневмония. Часто поражаются глаза (двусторонняя миотоническая катаракта). Характерны эндокринные расстройства (нарушение менструального цикла, гипогонадизм у мужчин, нарушения репродуктивной функции, сахарный диабет 2-го типа). Могут иметь место нарушение функции поджелудочной/щитовидной железы, половых желез, гипоталамуса.
Одним из частых проявлений является облысение в лобно-теменной области. Облысение сочетается с изменением структуры волос. Волосы становятся ломкими, тусклыми на фоне повышенной ломкости/тусклости волос. Зачастую страдает функция ЖКТ (дисфагии/нарушения перистальтики), отмечается прогрессирующий сколиоз, появляются костные деформации лобных костей, грудной клетки, стоп.
У детей при врожденной форме миотонической дистрофии отмечается выраженная мышечная гипотония с поражением мимической, жевательной и глазодвигательных мышц (симптомокомплекс «вялого ребенка»).
Анализы и диагностика
Для диагностики используются самые различные методы, начиная от молекулярной и генетической до различных инструментальных (УЗИ. МРТ, КТ, электрокардиоргамма/эхокардиография, электромиография, биопсия мышц и др.) и лабораторных методов исследования в зависимости от заболевания, протекающего с дистрофическими проявлениями.
Лечение
Лечение болезней, протекающих с явлениями дистрофии определяется конкретным заболеванием. Рассмотри лишь уже рассмотренные заболевания.
Лечение атаксии Фридрейха
Полного выздоровления не наступает, а основное лечение сводится к замедлению его прогрессирования и снижению риска развития ряда симптомов/осложнений. С этой целью показаны антиоксиданты, препараты митохондриального ряда и другие лекарственные средства, уменьшающие аккумуляцию в митохондриях железа и влияющие на различные уровни энергетического метаболизма. Рекомендуется назначение одновременно как трех и более лекарственных средств из этих групп: антиоксиданты — витамины А и Е, Глицин, Идебенон (синтетический аналог коэнзима Q 10), который эффективно тормозит развитие нейродегенеративного процесса, а также, развитие гипертрофической кардиомиопатии. Также назначаются кофакторы энзимных реакций и препараты, способствующие повышению активности митохондрий (Кокарбоксилаза, Нивалин, Прозерин, Оксазил, Нейромидин, Галантамин, Рибоксин, Предуктал, 5-гидроксипрофан).
Проводится общеукрепляющая терапия (витамины), а терапия, направленная на нормализацию тканевого метаболизма (Пирацетам, Тиоцетам, Церебролизин, Метионин, Ретаболил, Аминалон, АТФ-лонг, Актовегин, Гинкго-билоба, Прозерин, Ацефен, Пиритинол, Никотиновая кислота) курсами. Для замедления дистрофических процессов могут назначаться глюкокортикоиды (Преднизолон, Оксазолон, Дефлазакорт). Показаны препараты с нейротрофическим эффектом (Церебрум композитум, Траумель).
Хороший эффект дает метаболическая терапия, улучшающая процессы метаболизма в клетках миокарда, костной ткани, печени, скелетных мышцах, способствующая нормализации жирового обмена (Левокарнитин, витамин Д3, препараты кальция, витамины группы В – Цианокобаламин/Тиамин-хлорид). Показано введение в спастичные мышцы Ботулотоксина. Дополнительно проводится лечение конкретно развившегося заболевания со стороны сердечно-сосудистой системы, диабета, катаракты, поджелудочной железы, искривлений позвоночного столба и др.
Лечение миотонической дистрофии
Какой-либо этиопатогенетической терапии миотонической дистрофии нет. Проводится преимущественно симптоматическое лечение, включающее в комплекс мероприятий при нервно-мышечных заболеваниях. Могут назначаться препараты, уменьшающие ригидность мышц (Мексилетин, Хинидин, Карбамазепин, Фенитоин, Прокаинамид и др.). Существенное облегчение приносит ортопедическая коррекция, физиотерапевтическое лечение. Для поддержания мышц назначается фармакопунктура с церебролизином, актовегином, нейромидином, цианокобаламином. При развитии остеопороза показаны кальцийсодержащие препараты, например, Кальцемин Адванс. При развитии конкретного заболевания проводится его лечение.



