Хирургическая коррекция пола

Хирургическая коррекция пола является, пожалуй, единственным возможным методом устранения расхождения между анатомическим полом пациента и его гендерной самоидентификацией. Данные вмешательства выполняются только при установленном и зафиксированном врачом-психиатром диагнозе «транссексуализм» и наличии решения врачебной комиссии о проведении медицинской коррекции пола.
Противопоказаниями к медицинской коррекции пола являются:
Первым этапом коррекции пола является заместительная гормональная терапия, которая приводит к определенным изменениям:
Этап приема гормональных препаратов при смене пола длится от 1 года и более, далее возможно решение вопроса о коррекции пола хирургическими методами.
Трансформация из мужчины в женщину
Наиболее часто выполняется хирургическая трансформация из мужчины в женщину. Процедура хирургической смены пола начинается с орхиэктомии и вагинопластики, далее выполняется феминизирующая маммопластика. По желанию пациента проводится удаление кадыка и ряд пластических операций на лице и теле.
Трансформация из женщины в мужчину
Технически более сложна операция трансформации из женщины в мужчину. Для того, чтобы сменить пол хирургически, необходимо сначала пройти операции по удалению груди и органов малого таза. Комплекс хирургической коррекции пола в данном случае может включать в себя:
Нередко хирургическая коррекция пола сопровождается рядом пластических операций на лице.
Научно-практический центр хирургии помогает пациентам скорректировать пол хирургически и обрести гендерную идентичность. Врачи-хирурги нашего центра обладают высоким уровнем профессионализма и большим практическим опытом проведения подобных трансформаций, что в сочетании с современным хирургическим оборудованием и комфортными условиями пребывания обеспечивает наилучшее качество оказываемых услуг.
Что такое пенальная инверсия
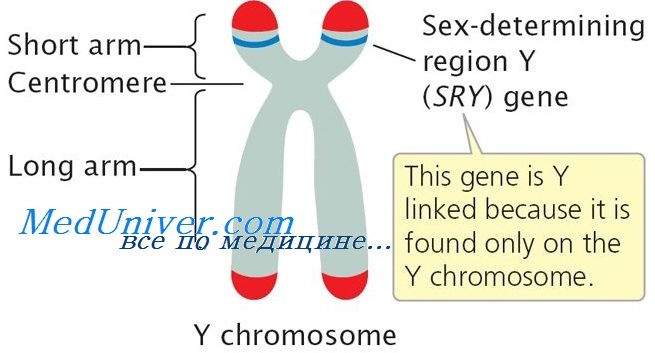
Этиология и встречаемость инверсии пола. Инверсия пола — панэтническое и генетически разнородное заболевание. Среди пациентов с полной гонадной дисгенезией наиболее частые причины инверсии пола — точковые мутации, делеции или транслокации гена SRY.
Приблизительно 80% мужчин 46,ХХ с полной дисгенезией гонад имеют транслокацию SRY на Х-хромосому, и 20-30% женщин 46,XY с полной дисгенезией гонад имеют мутацию или делецию гена SRY. Встречаемость мужчин 46,ХХ и женщин 46,XY составляет примерно 1 на 20 000.
Патогенез инверсии пола
SRY — связанный с ДНК белок, изменяющий структуру хроматина, изгибая ДНК. Эта связь и изменение свойств ДНК указывают, что белок SRY регулирует экспрессию генов.
В ходе нормального развития человека SRY необходим для образования мужских половых органов, но для образования женских половых органов его отсутствие не обязательно. Точный механизм влияния SRY на развитие мужских гонад неопределен, хотя некоторые наблюдения указывают, что SRY подавляет отрицательный регулятор развития тестикул.
Мутации в гене SRY, выявляемые у XY-женщин, вызывают потерю его функции. Среди XY-женщин 10-15% имеют делецию SRY (женщины XY, SRY), еще 10-15% имеют точковые мутации в пределах гена. Точковые мутации в гене SRY нарушают связь с ДНК или поворот ДНК.
Нарушение SRY у мужчин XX — транслокация SRY с Yp на Хр (мужчины XX, SRY+; рис. С-36, см. цв. вклейку). В мужском мейозе происходит кроссинговер между псевдоаутосомными регионами Хр и Yp; этот кроссинговер обеспечивает правильное расхождение и соответствие последовательностей в псевдоаутосомных регионах X и Y.
Тем не менее иногда рекомбинация происходит неправильно, захватывая центромеру, что приводит в передаче специфических последовательностей короткого плеча Yp-, включая ген SRY, на короткое плечо Хр.
Кроме SRY, Y-хромосома содержит, по крайней мере, три локуса (локусы факторов азооспермии AZFa, AZFb и AZFc), необходимых для нормального сперматогенеза. Отсутствие этих локусов, по крайней мере, частично, объясняет бесплодие у мужчин XX, SRY+.
Х-хромосома также содержит несколько локусов, необходимых для функционирования яичников и женской фертильности. Для развития овоцитов достаточно наличия одной Х-хромосомы, но для использования овоцитов необходимы две Х-хромосомы. В соответствии с этим наблюдением, у женских плодов XY овоциты закладываются, но фолликулы вырождаются еще до рождения или вскоре после него. Следовательно, бесплодие женщин XY объясняется отсутствием второй Х-хромосомы.
Фенотип и развитие инверсии пола
Мужчины XX, SRY+ имеют много признаков синдрома Кляйнфелтера (47,XXY), включая гипогонадизм, азооспермию, гиалиноз семявыносящих канальцев и гинекомастию. Несмотря на сниженный синтез тестостерона, у большинства пациентов пубертат развивается самостоятельно, хотя для достижения полной вирилизации может потребоваться введение тестостерона.
В отличие от пациентов с синдромом Кляйнфелтера, большинство мужчин 46,ХХ имеют нормальный рост, нормальные пропорции скелета, интеллект и меньше психосоциальных проблем. Пациенты с большим участком Yp в Х-хромосоме имеют большее сходство с синдромом Кляйнфелтера.
Женщины XY, SRY- имеют полную дисгенезию гонад, они обычно выше среднего роста для нормальных женщин. Эти пациентки имеют физические характеристики синдрома Тернера, только если делеция SRY связана с обширной делецией Yp. Поскольку у этих пациенток вместо яичников присутствуют только рубчики (полоски соединительной ткани), спонтанно пубертат у них не развивается.
В отличие от полной пенетрантности и сравнительно однородной экспрессивности, наблюдаемых при транслокациях или делециях гена SRY, точковые мутации SRY проявляют как неполную пенетрантность, так и варьирующую экспрессивность. Пациенты с точковыми мутациями в гене SRY обычно имеют полную дисгенезию гонад, рост выше среднего и у них не развиваются вторичные половые признаки.
Тем не менее описано несколько точковых мутаций SRY, связанных с бесплодием (полной дисгенезией гонад) при женском фенотипе и фертильностью при мужском фенотипе в одной и той же семье.
Особенности фенотипических проявлений инверсии пола:
• Возраст начала: пренатальный
• Бесплодие
• Недоразвитие вторичных половых признаков
• Однозначные гениталии
Лечение инверсии пола
У пациентов с полной дисгенезией гонад диагноз инверсии пола обычно устанавливают при расхождении данных УЗИ плода и кариотипа плода, или из-за отсутствия или неполного развития вторичных половых признаков и бесплодия. Доказательство того, что инверсия пола вторична по отношению к аномалии экспрессии SRY, требует демонстрации соответствующего изменения в гене SRY.
Для мужчин XX, SRY+ назначение андрогенов обычно способствует вирилизации, но лечение, предохраняющее от азооспермии, в настоящее время невозможно. Введение дополнительных андрогенов не помогает избежать гинекомастии. Если гинекомастия становится достаточно выраженной, пациентам приходится прибегать к хирургическому лечению.
Для женщин XY, SRY+ и женщин XY с точковыми мутациями гена SRY терапия эстрогенами обычно начинается в возрасте 14-15 лет, чтобы обеспечить развитие вторичных половых признаков. Для вызывания менструаций в схему лечения добавляют прогестерон либо в момент первого вагинального кровотечения, либо через 2 года после начала лечения эстрогенами.
Кроме того, из-за риска развития опухолей гонад, рекомендуется удаление неполноценных яичников после завершения роста скелета.
Как и при всех нарушениях половой дифференцировки или несоответствиях генетического и фенотипического пола, чрезвычайно важны психосоциальная коррекция и консультирование семьи и пациента. Большинство семей и пациентов имеют затруднения в понимании медицинских данных и получении соответствующих психосоциальных установок.
Риски наследования инверсии пола
Неправильная рекомбинация de novo — наиболее частая причина появления мужчин XX, SRY+ и женщин XY SRY_; следовательно, большинство пар с больным ребенком имеет низкий риск повторения у будущих детей. Тем не менее, иногда мужчины XX SRY+ и женщины XY SRY возникают в результате наследуемой делеции SRY или транслокации от отца со сбалансированной транслокацией между Хр и Yp.
Если отец — носитель транслокации, все дети будут или мальчиками XX, SRY+ или девочками XY, SRY-. Поскольку мужчины XX, SRY+ и женщины XY,SRY всегда бесплодны, они не могут передать заболевание потомкам.
Большинство женщин XY с точковыми мутациями в гене SRY имеют новые мутации. Родители больного ребенка, следовательно, обычно имеют низкий риск повторения у будущих детей; тем не менее, поскольку некоторые мутации SRY имеют неполную пенетрантность, здоровые фертильные отцы могут нести мутации SRY, способные вызвать инверсию пола у их детей с генотипом XY.
Пример инверсии пола. Женщина, 37-летняя служащая, беременна первым ребенком. Из-за возрастного риска иметь ребенка с хромосомной аномалией она решила провести амниоцентез, чтобы оценить кариотип плода; результат кариотипа нормальный, 46.ХХ. Тем не менее на 18-й нед гестации при УЗИ обнаружен нормальный плод мужского пола; последующее подробное УЗИ подтвердило мужской пол плода.
Женщина была здорова перед и в течение беременности, без влияния инфекций или лекAPCтв в ходе беременности. Ни она, ни ее партнер не имели в семейном анамнезе случаев неоднозначных половых органов, бесплодия или врожденных пороков.
Повторный хромосомный анализ подтвердил нормальный кариотип 46.ХХ, но методом FISH выявлено присутствие сигнала гена региона половой детерминации Y (SRY) в одной из Х-хромосом. На 38-й нед беременности пациентка без осложнений самостоятельно родила фенотипически нормального ребенка мужского пола.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Инверсия пола 46 XY
OMIM 400044
Наша команда профессионалов ответит на ваши вопросы
Инверсия пола, 46,XY
Наличие женского фенотипа при нормальном мужском кариотипе характеризует XY-инверсию пола. Наиболее частой причиной данного нарушения формирования пола является синдром Свайера – это полная или «чистая» дисгенезия гонад при кариотипе 46,XY. Частота XY-дисгенезии гонад составляет 1 на 30000 человек. Больные имеют женский фенотип без признаков двойственности полового развития: феминное телосложение, развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку и маточные (фаллопиевы) трубы. Однако у пациентов с синдромом Свайера практически отсутствуют женские половые железы, которые в данном случае представлены дисгенетичными гонадами, представляющими собой соединительнотканные тяжи (стреки) с небольшими включениями железистой ткани, овариально-подобной стромы без фолликулов. Как правило, диагностирование синдрома Свайера происходит у девочек в пубертатный период, когда у них не происходит нормального полового развития. Причиной обращения к врачу при этом является задержка полового развития и отсутствие начала менструаций, реже наличие злокачественных новообразований, происходящих из дисгенетичных гонад. Так как дисгенетичные гонады подвержены озлокачествлению, показано их удаление в детстве или на момент постановки диагноза XY-дисгенезии гонад. После оперативного лечения пациенткам, как правило, еще в подростковом возрасте назначается заместительная гормональная терапия, чтобы достичь нормального развития вторичных половых признаков и предотвратить развитие остеопороза. У женщин с XY-дисгенезией гонад нет собственных яйцеклеток, однако в некоторых случаях она в состоянии выносить плод, полученный в программе ЭКО при оплодотворении донорской яйцеклетки сперматозоидами супруга.
Инверсия пола, 46,XY тип 1 (OMIM 400044)
Наиболее частой из известных причин «чистой» формы дисгенезии гонад 46,XY являются микроструктурные перестройки Y-хромосомы c утратой гена SRY (Sex-determining region Y), а также точковые мутации данного гена. У 10-15% больных с синдромом Свайера обнаруживают отсутствие локуса SRY. В большинстве случаев это обусловлено утратой фрагмента дистальной части короткого плеча Y-хромосомы (Yp11.3), вследствие X-Y транслокации. Еще у 10-15% пациентов с данным синдромом выявляют мутации гена SRY.
Ген SRY локализован на коротком плече Y хромосомы и кодирует транскрипционный фактор – белок, связывающийся с генами, определяющими развитие пола плода по мужскому типу. Мутации в гене SRY приводят к синтезу функционально неполноценного белка и к нарушению дифференцировки клеток Сертоли и формирования семенных канальцев в развивающихся бипотенциальных гонадах плода, что вызывает дисгенезию гонад и развитие остальных органов половой системы по женскому типу, несмотря на наличие Y-хромосомы в кариотипе.
Инверсия пола, 46,XYтип 2 (OMIM 300018)
Данный тип XY-инверсии пола обусловлен дупликаций гена NR0B1 (DAX-1). Ген NR0B1локализован на коротком плече Х хромосомы (локус Хp21.3). Кодируемый этим геном белок DAX-1 играет важную роль в развитии и функции некоторых органов эндокринной системы, в том числе и половых желез. Еще внутриутробно он контролирует активность генов, участвующих в формировании этих тканей, а в постнатальном периоде DAX-1 регулирует выработку в них гормонов. Белок DAX-1 оказывает дозо-зависимый эффект на органы эндокринной системы. Дупликация гена NR0B1, а также делеция располагающегося рядом с геном NR0B1 локуса, негативно-регулирующего его транскрипцию приводит к XY-инверсии пола, обусловленной XY-дисгенезией гонад часто сочетающейся с нарушением функции надпочечников. Точковые мутации этого гена у пациентов с кариотипом 46,XY вызывают нарушение развития тестикулярной ткани, приводят к дефициту маскулинизации. Мутации в этом гене также вызывают Х-сцепленную гипоплазию надпочечников, как у пациентов с кариотипом 46,ХХ так и 46,XY.
Инверсия пола, 46,XY тип 3 (OMIM 612965)
Инверсия пола, 46,XY тип 4 (OMIM 154230)
Эта форма XY-инверсии пола обусловлена делецией локуса 9p24.3. У пациенток отмечают нормально развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку, при гистологическом исследовании гонад обнаруживают наличие незрелой тестикулярной ткани, содержащей клетки Сертолли, и отсутствие зрелых половых клеток. Инверсия пола у данных пациентов, вероятно, обусловлена потерей одной из копий дозо-чувствительного гена, локализованного в данном локусе. Генами-кандидатами являются DMRT1 и DMRT2.
Инверсия пола, 46,XY тип 5 (OMIM 613080)
Данная аутосомно-рецессивная форма инверсии 46,XY обусловлена наличием мутаций в гене CBX2, расположенного на хромосоме 17 (локус 17q25). В 2009 году Байсон-Лаубер описал случай новорожденной девочки с кариотипом 46,XY, у которой в результате секвенированияв гене CBX2 были обнаружены две мутации (P98L и R443P). В результате исследований у девочки были обнаружены нормально развитые яичники, с наличием овариальной ткани и первичных фолликулов, а также влагалище и матка. Однако возраст еще был слишком мал, чтобы оценить ее фертильность и дальнейшее половое развитие.
Инверсия пола, 46,XY тип 6 (OMIM 613762)
XY-инверсия пола связана с наличием мутации в гетерозиготном состоянии в гене MAP3K1, расположенном в локусе 5q11.2. Пациентки с данной формой дисгенезии гонад имеют высокий рост, который, вероятно, обусловлен избыточной продукцией андрогенов, тяжевидные яичники, гипоплазированную матку, иногда наблюдается клиторомегалия.
Инверсия пола, 46,XY тип 7 (OMIM 233420)
Инверсия пола обусловлена наличием у пациенток мутаций в гомозиготном или компаунд-гетерозиготном состоянии в гене DHH, расположенного в локусе 12q13.12. У нескольких пациенток было описано наличие недоразвитой матки, также присутствовали фаллопиевы трубы и наблюдали полную форму ХY-дисгенезии гонад (тяжевидные гонады, которые часто озлокачествлялись).
Инверсия пола, 46,XYтип 8 (OMIM 614279)
Данный тип XY-инверсии пола обусловлен мутациями гена AKR1C2, лежащего в локусе 10p15, отвечающего за альтернативный путь синтеза дигидротестостерона. Мутации сцепленного с ним гена AKR1C4, который сегрегирует вместе с геном AKR1C2, могут влиять на выраженность фенотипических проявлений.
В Центре Молекулярной Генетики проводится молекулярный анализ ключевых генов, контролирующих дифференцировку пола, в частности выполняется секвенирование генов SRY и NR5A1 (SF1), а также с помощью количественного метода MLPA проводится поиск делеций и дупликаций генов SRY, NR5A1 (SF1), NR0B1 (DAX-1).
Заболевания по направлению Синдром карпального канала
Тел.: 8-800-25-03-03-2
(бесплатно для звонков из регионов России)
Санкт-Петербург, наб. реки Фонтанки, д. 154
Тел.: +7 (812) 676-25-25
Санкт-Петербург, В.О., Кадетская линия, д. 13-15
Тел.: +7 (812) 676-25-25
Санкт-Петербург, ул. Циолковского, д.3
Тел.: +7 (812) 676-25-10
Синдром запястного канала (карпальный туннельный синдром) — это заболевание, выражающееся в защемлении и воспалении срединного нерва. Проявляется в нарушении чувствительности рук и постепенной утрате двигательной активности. Причиной болезни является сдавливание срединного нерва в анатомическом канале.
Описание заболевания
Через запястный (карпальный) канал проходят сосуды, нервы, синовиальные оболочки, сухожилия. Здесь же располагается срединный нерв — главный нерв руки, идущий от плечевого сплетения до кончиков пальцев. Отвечает за координирование движения, мелкую моторику рук, сужение и расширение кровеносных сосудов от действия внешних раздражителей, регулирует работу потовых желез. Ширина канала ограничена с трех сторон костями и поперечной связкой.
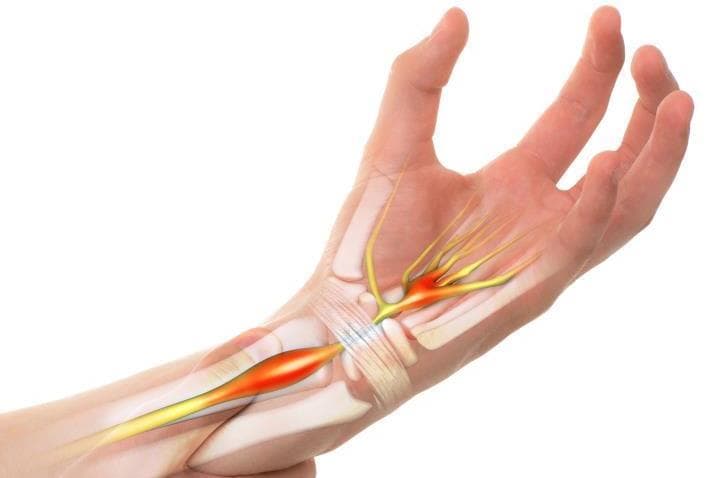
Запястный канала сам по себе довольно узок, что способствует образованию различных патологий. Любое дополнительное сужение влечет за собой сдавливание отростков нервных волокон и сосудов, нарушается кровоснабжение. Патологические процессы протекают медленно, начинаются с потери физиологической чувствительности, приводят к моторным, трофическим расстройствам. Картина заболевания имеет четкую симптоматику с характерной клиникой, что позволяет легко поставить диагноз даже на начальном этапе.
Симптомы заболевания
1. Снижение чувствительности кисти. Сначала пациенты начинают замечать онемение пальцев рук сразу после пробуждения. Со временем периоды парестезии увеличивается, к нему добавляются чувства жжения, боли, холода либо жара. Для синдрома запястного канала характерно одновременное, кардинально противоположное изменение чувствительности. На одном участке она повышается, но другая часть ладони ничего не чувствует.
2. Жгучая боль в ладонях с прострелом в пальцы. Первоначально боль ощущают в зоне защемления нерва, затем она постепенно начинает затрагивать всю руку: от плеча до кончиков пальце. Это сопровождается опуханием запястья. Как правило, болит доминантная рука: правая у правши, левая у левши, но встречаются и двусторонние симптомы.
3. Мышечная слабость и нарушение двигательной активности. В результате длительного защемления и воспаления нервных волокон кисть теряет мышечную силу, появляется ограничение в движении. Становится трудно управлять мелкими предметами, например, застегивать пуговицы. Ослабевает функция хватания, а затем и удерживания предметов. Постепенно снижение работоспособности руки приводит к атрофии мышц и деформации кисти.
4. Поражения вегетативной нервной системы. Это выражается в бледном, а иногда синюшном цвете руки, сухости кожи, ломкости ногтей, похолодание кисти.
Естественно, синдром запястного канала влияет на общее физическое и психическое состояние больного. Он теряет трудоспособность, снижается качество жизни. Регулярные боли и постоянное ощущение дискомфорта усиливаются по ночам, что мешает нормальному сну, вызывает бессонницу.
Характерным для этого заболевания является то, что снять неприятные ощущения больной может самостоятельно, не прибегая к медикаментам. Для этого достаточно опустить руки вниз, подвигать пальцами, встряхнуть кисти, размять их, сделать массаж, гимнастику. Эти манипуляции помогают восстановить кровоснабжение, но, к сожалению, ненадолго, поскольку не избавляют от причин.
Диагностика
Поставить диагноз при этом заболевании легко, достаточно опросить пациента и провести несколько тестов, поскольку синдром запястного канала обладает яркими отличительными признаками. Среди них описанные выше симптомы и сохранение функций мизинца, который при защемлении нерва и сосудов в карпальном канале не поражается.
Так же врач проводит пальпацию и следующие тесты:
Тест Хоффмана — Тинеля: простукивание области срединного нерва. При карпальнотуннельной патологии пациент ощущает ладонью онемение, покалывание, жжение.
Тест Фалена: максимально согнув кисть в запястном суставе, пациент чувствует боль и онемение ладони.
Поднятые над головой руки сложно удержать дольше 1 минуты, начинается болевой синдром и онемение конечностей.
Невозможно соединить (соприкоснуть) большой палец с мизинцем.
Для подтверждения правильности диагноза, определения степени поражения и сопутствующих осложнений, назначают дополнительные обследования:
Электронейромиография для оценить проводимость нервных импульсов- золотой стандарт диагностики туннельного синдрома.
Ультрасонография помогает обнаружить повреждения и воспаления.
Поставить диагноз и проводить лечение могут врач-невролог, физиотерапевт и кистевой хирург.
Лечение синдрома запястного канала
Все проводимые мероприятия направлены на устранение причины заболевания — полное или частичное устранение сжатия срединного нерва. Методика терапии зависит от степени тяжести синдрома. Выбирают между консервативным и хирургическим лечением. Также в задачи врача входит выявление причины возникновения заболевания для того, чтобы ограничить движения, ухудшающие симптоматику.
1. Консервативное лечение
Пациентам назначают ношение ортеза, удерживающее сустав запястья в безопасном для сосудов и нервов положении. Его рекомендуется носить постоянно, в дневное время и ночью. Согласно статистическим данным ортезирование эффективно на ранней и средней стадии развития болезни. Рекомендуется перейти на бессолевую диету, ограничить употребление жидкости, чтобы снизить отечность.
Назначают и медикаменты:
Нестероидные противовоспалительные агенты для снижения болевых ощущений и воспаления.
Кортикостероиды перорально, либо инъекционно для купирования боли и восстановления подвижности суставов.
Сосудистые препараты для улучшения кровоснабжения тканей.
Диуретики для снятия отечности.
Витамин B6 для облегчения симптоматики и увеличения эффективности лечения.
Ускорить процесс терапии, быстрее достигнуть положительного результата помогает физиотерапия: электрофорез, УВЧ, магнитотерапия и другие методы. Часто назначают курс лечебной физкультуры и массаж. Комплексное сочетание этих способов терапии и употребление медикаментов приводит к ремиссии у 59% пациентов. Остальные вынуждены в среднем через 1-2 лет обратиться к кистевому хирургу для проведения операции.
2. Хирургическое лечение
При неэффективности консервативного лечения и при запущенности заболевания назначается проведение хирургической операции. Занимаются ими только кистевые хирурги — узкопрофильные специалисты, проводящие оперативные пособия на кисти. Цель операции —освободить срединный нерв, уменьшить давление. Существует два способа раскрытия запястного канала:
Открытое хирургическое вмешательство, при котором выполняют разрез на запястье длиною около 3 см. Получив доступ, хирург рассекает связку, увеличивая объём запястного тоннеля.
Эндоскопия отличается меньшей длиной разреза, которая составляет до 1,5 см, но проводится в двух местах. Они необходимы для ввода микрокамеры, используя которую можно провести рассечение связки.
Оба варианта хирургии проводят под местной анестезией. Иногда требуется иссечение тканей вокруг нерва и сухожилий, имеющие рубцовые изменения, завязи и тяж. В среднем, процедура проводится в течение 20 мин.
Первое время пациент может чувствовать боль в области рубцов и физическую слабость запястья. До полного восстановления потребуется несколько месяцев, срок индивидуален в каждом конкретном случае. Реабилитация состоит из гимнастики, массажа, физиотерапии. На период восстановления многим приходится отказаться от своего вида деятельности, если он влияет на восстановление.
По статистике до 90% пациентов прошедших через хирургическое лечение полностью восстанавливают физическую активность кистей и избавляются от симптомов синдрома запястного канала. Рецидивы встречаются в 8 — 12% случаев.
3. Альтернативные методы лечения.
Можно снять симптомы туннельного синдрома без медикаментов и операций. Для этого необходимо:
Снизить нагрузку на кисти рук и запястья, исключить деятельность, ухудшающую состояние.
Зафиксировать руку шиной, ортезом или повязкой из эластичного бинта для ограничения подвижности.
Снять отечность при помощи льда, ограничить употребление жидкости и соли.
Выполнять гимнастику рук, направленную на растяжение суставов. Рекомендуем упражняться под присмотром физиотерапевта.
Если улучшения не наступает, то не стоит тянуть с обращением к врачу.
Что будет, если не лечиться?
Синдром карпального канала не угрожает жизни, представляет собой медленно текущий патологический процесс. Заболевание имеет благоприятный прогноз выздоровления, но из-за пренебрежительного отношения к своему здоровью, некоторые люди не спешат посещать врача. Со временем это приводит к снижению двигательной активности, силе мышц, нарушению мелкой моторики. Если не лечиться, начнутся необратимые процессы, срединный нерв атрофируется. Это приведет утрате функциональности кисти, вернуть которую уже будет невозможно.
Причины развития синдрома
Заболевание карпально-туннельного синдром развивается под воздействием нескольких факторов. Уменьшение диаметра запястного канала может происходить из-за сдавливания нерва. Отека окружающих тканей и по обеим причинам одновременно. Первопричиной является травматизация, как единовременная, так и микротравмы наносимые регулярно из-за образа жизни либо трудовой деятельности.
1. «Опасные» профессии. Среди профессий сходящий в зону риска:



