Что такое равновесная концентрация лекарственного препарата
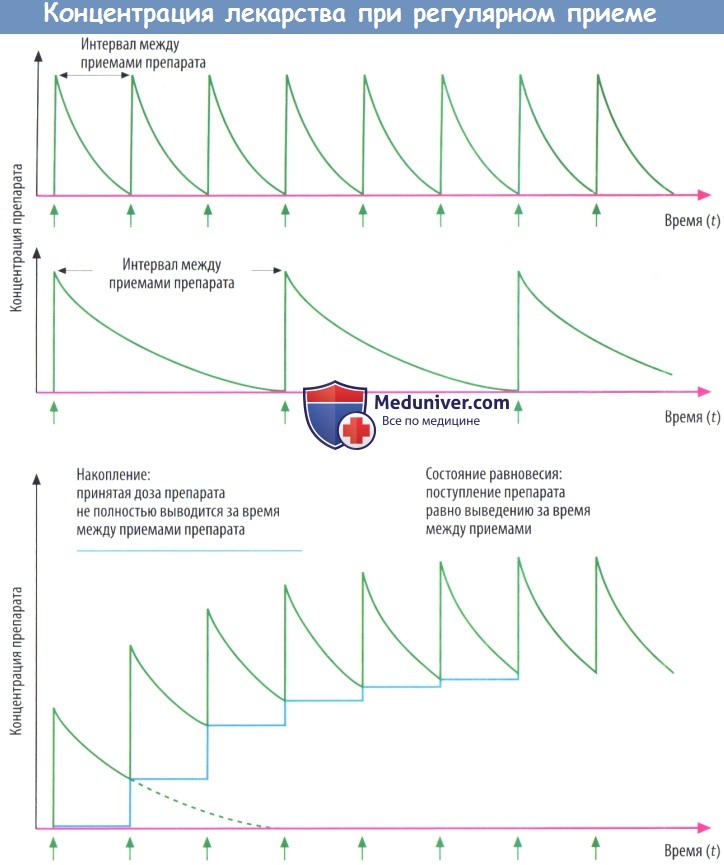
а) Концентрация препарата в крови при повторном введении. Если больной принимает препарат в течение длительного периода через одинаковые промежутки времени, подъем и падение концентрации лекарственного вещества в крови определяется взаимоотношением между t1/2 и временным интервалом между дозами.
В том случае, если количество препарата, поступившего в организм, элиминируется прежде, чем принята следующая доза, концентрация вещества при повторных приемах через равные промежутки времени будет постоянной.
Если препарат поступает в организм прежде, чем предыдущая доза полностью элиминируется, следующая доза «добавляется» к остаточному количеству препарата, все еще присутствующему в организме, т. е. вещество накапливается.
Чем короче интервал между дозами по отношению к t1/2, тем выше остаточное количество препарата, к которому прибавляется очередная доза, и тем активнее препарат накапливается в организме.
Однако при фиксированном интервале межу приемами препарат не накапливается бесконечно, в итоге наступает состояние равновесия (концентрация Css), или равновесное накопление. Так происходит потому, что активность процессов элиминации зависит от концентрации. Чем выше концентрация препарата, тем большее его количество элиминируется за единицу времени.
После приема нескольких доз концентрация достигает уровня, при котором количество препарата, выведенного и поступившего в организм за единицувремени, становится одинаковым, т. е. достигается состояние равновесия.
В пределах этого диапазона концентрации уровень вещества в крови продолжает повышаться (пик) и падать (низшая точка) в процессе приема лекарственного средства через равные промежутки времени.
Максимальное значение равновесной концентрации (Css) зависит от количества препарата (D), принятого за время между введениями (τ), и клиренса (Cl): C55 = D/(τ x Cl).
Скорость, с которой достигается равновесное состояние, соответствует скорости элиминации препарата. Время, необходимое для достижения 90%-го плато концентрации, примерно в 3 раза превышает t1/2 элиминации.
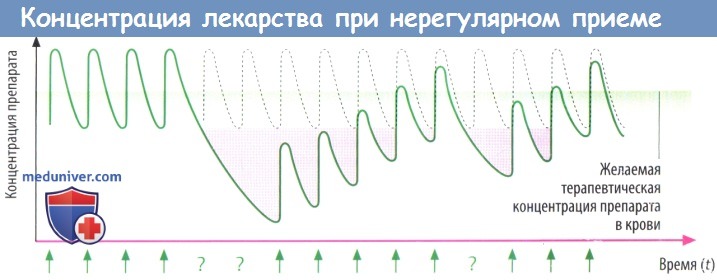
б) Концентрация препарата в крови при приеме через неравные промежутки времени. На практике бывает трудно добиться такого уровня препарата в крови, который колебался бы равномерно около желаемой эффективной концентрации. Например, если пропущены две дозы подряд, концентрация препарата в крови падает ниже терапевтической, и требуется больше времени, чтобы она вновь повысилась до желаемого уровня.
Нередко пациенты не соблюдают режим приема препарата (приверженность к лечению — строгое выполнение рекомендаций врача).
В результате концентрация препарата в крови в предутренние часы может опускаться ниже желаемой, а порой и крайне необходимой.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
ОСНОВНЫЕ ВОПРОСЫ ФАРМАКОКИНЕТИКИ




![]()
![]()
Фармакокинетика — раздел клинической фармакологии, предметом которого является изучение процессов всасывания, распределения, связывания с белками, биотрансформации и выведения лекарственных веществ. Ее развитие стало возможным благодаря разработке и внедрению в практику высокочувствительных методов определения содержания лекарственных веществ в биологических средах — газожидкостной хроматографии, радиоиммунных, ферментно-химических и других методов, а также благодаря разработке методов математического моделирования фармакокинетических процессов. На основании данных о фармакокинетике того или иного препарата определяют дозы, оптимальный путь введения, режим применения препарата и продолжительность лечения. Регулярный контроль содержания лекарственных средств в биологических жидкостях позволяет своевременно корригировать лечение.
Фармакокинетические исследования необходимы при разработке новых препаратов, их лекарственных форм, а также при экспериментальных и клинических испытаниях ЛС.
Процессы, происходящие с лекарственными препаратами в организме, могут быть описаны с помощью ряда параметров.
Одним из основных показателей, определяющих фармакологический эффект, считают концентрацию ЛС на уровне рецептора, однако в условиях целостного организма установить её невозможно. В эксперименте было доказано, что в большинстве случаев между концентрацией препарата в крови и его содержанием в области рецептора существует корреляция.
В связи с этим для определения фармакокинетических параметров изучают содержание ЛС в крови. Для того чтобы получить соответствующее представление о поступлении препарата в кровь и выведении его из организма, отслеживают изменения концентрации ЛС в плазме крови на протяжении длительного времени. Содержание препаратов в плазме крови определяют методами жидкостной или газожидкостной хроматографии, с помощью радиоиммунного или иммуноферментного анализа и другими способами.
Такой график носит название фармакокинетической кривой (рис. 1).
 Время после введения
Время после введения
Константы скорости элиминации (Кel), абсорбции (Ка) и экскреции (Кex) – характеризуют соответственно скорость исчезновения препарата из организма путем биотрансформации и выведения, скорость поступления его из места введения в кровь и скорость выведения с мочой, калом, слюной и др.
Период полувыведения (Т1/2) — время, необходимое для уменьшения вдвое концентрации препарата в крови, зависит от константы скорости элиминации (Т1/2= 0,693/Кel).
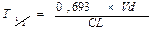
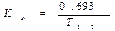
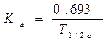
Распределение препарата в организме характеризуют период полураспределения, кажущаяся начальная и стационарная (равновесная) концентрации, объем распределения.
Период полураспределения (Т1/2,a) — время, необходимое для достижения концентрации препарата в крови, равной 50% от равновесной, т.е. при наличии равновесия между кровью и тканями.
Кажущаяся начальная концентрация (С0) — концентрация препарата, которая была бы достигнута в плазме крови при внутривенном его введении и мгновенном распределении по органам и тканям.
Равновесная концентрация (Сss) — концентрация препарата, которая установится в плазме (сыворотке) крови при поступлении препарата в организм с постоянной скоростью. При прерывистом введении (приеме) препарата через одинаковые промежутки времени в одинаковых дозах выделяют максимальную (Сssmax) и минимальную (Сssmin) равновесные концентрации.

Общий клиренс препарата (Clt) характеризует скорость “очищения” организма от лекарственного препарата.
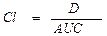 где Сl – общий клиренс; D – доза введенного препарата; AUC – площадь под фармакокинетической кривой.Выделяют почечный (Clr) и внепочечный (Cler) клиренсы, которые отражают выведение лекарственного вещества соответственно с мочой и другими путями (прежде всего с желчью). Общий клиренс является суммой почечного и внепочечного клиренса.
где Сl – общий клиренс; D – доза введенного препарата; AUC – площадь под фармакокинетической кривой.Выделяют почечный (Clr) и внепочечный (Cler) клиренсы, которые отражают выведение лекарственного вещества соответственно с мочой и другими путями (прежде всего с желчью). Общий клиренс является суммой почечного и внепочечного клиренса. 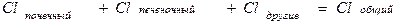
Абсолютная биодоступность (f) — часть дозы препарата (в %), которая достигла системного кровотока после внесосудистого введения, равна отношению AUC после введения исследуемым методом (внутрь, в мышцу и др.) к AUC после внутривенного введения. Относительную биодоступность определяют для сравнения биодоступности двух лекарственных форм для внесосудистого введения. Она равна отношению (AUC’/AUC)(D/D’) после введения двух сравниваемых форм. Общая биодоступность — часть принятой внутрь дозы препарата, которая достигла системного кровотока в неизмененном виде и в виде метаболитов, образовавшихся в процессе всасывания в результате так называемого пресистемного метаболизма, или “эффекта первичного прохождения”.
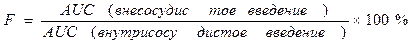
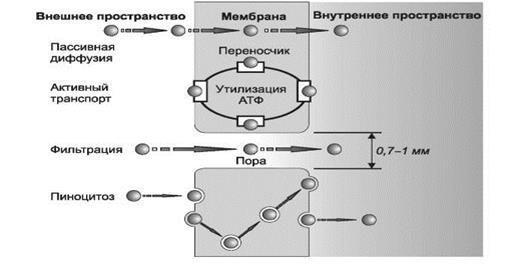
ВСАСЫВАНИЕ — процесс поступления лекарственного вещества из места введения в кровь. Существуют четыре механизма всасывания ЛС при энтеральном введении (рис. 2):
Ø пассивная диффузия;
Ø активный транспорт;
Ø фильтрация через поры;
Прохождение большинства лекарственных препаратов через слизистую оболочку пищеварительного тракта определяется их растворимостью в липидах и ионизацией. При приеме лекарственных веществ внутрь скорость их абсорбции отличается в различных отделах ЖКТ.
После прохождения через стенку желудка и/или кишечника лекарственный препарат поступает в печень. Некоторые лекарственные вещества под влиянием ферментов печени подвергаются значительным изменениям (“эффект первичного прохождения”). Именно поэтому, а не вследствие плохой абсорбции, для достижения достаточного эффекта дозы некоторых препаратов (пропранолола, аминазина, опиатов) при приеме их внутрь должны быть значительно больше, чем при внутривенном введении. Биотрансформацию вещества при первичном прохождении через печень в процессе всасывания называют пресистемным метаболизмом. Интенсивность пресистемного метаболизма зависит от скорости тока крови в печени.
На процесс всасывания лекарств в желудке и кишечнике оказывает влияние рН, который в желудке равен 1-3, в двенадцатиперстной кишке — 5-6, а в тонкой и толстой кишках — около 8. Кислоты легче всасываются в желудке, а основания — в тонкой или толстой кишке.
Под действием кислой среды желудка некоторые лекарственные средства, в частности бензилпенициллин, могут разрушаться.
На лекарственные препараты оказывают также действие ферменты желудочно-кишечного тракта, которые способны инактивировать белки и полипептиды (АКТГ, вазопрессин, инсулин и т.д.), а также некоторые другие вещества (прогестерон, тестостерон, альдостерон). Соли желчных кислот в свою очередь могут ускорить всасывание лекарственных средств или замедлить его при образовании нерастворимых соединений.
На всасывание лекарственных веществ влияют также моторика желудочно-кишечного тракта, объем и состав пищи, количество принимаемой жидкости, интервал времени между едой и приемом препаратов. Так, молоко нарушает всасывание тетрациклинов, ампициллина и амоксициллина. Следует учитывать и стимулирующее действие пищи на секрецию желудочного сока и соляной кислоты.
Для переноса веществ в ЖКТ особое значение имеют большая площадь поверхности кишечника и влияние постоянного кровотока в слизистой оболочке на градиенты концентрации между просветом кишечника и кровью. Путем диффузии и осмоса через слизистую оболочку кишечника переносятся, в частности, вода, С1 ¯, а также такие вещества, как аскорбиновая кислота, пиридоксин и рибофлавин. Поскольку клеточные мембраны содержат большое количество липидов, для диффузии через мембрану вещества должны быть в некоторой степени жирорастворимыми. Согласно теории неионной диффузии, указанным путем переносятся главным образом недиссоциированные соли слабых кислот или слабых оснований. Это необходимо учитывать при назначении лекарств, большая часть которых всасывается путем диффузии. Для переноса какого-либо вещества в соответствии с уравнением Гендерсона-Гассельбаха особое значение имеет рКа этого вещества и рН в просвете кишечника:
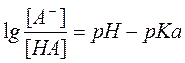 ,
, 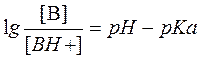 , где
, где
[А¯], [ВН + ] – молярные концентрации ионизированных,
[НА], [В] – неионизированных форм кислоты НА и основы В;
рН – кислотно-основной показатель среды;
рКа – логарифм константы диссоциации соединения, количественно равный значению рН, при котором анализируемое соединение диссоциирует наполовину.
Таким образом, факторы, влияющие на процессы всасывания ЛВ, разнообразны: растворимость вещества в липидах, степень ионизации молекулы (чем меньше ионизированная молекула, тем лучше она всасывается), перистальтика кишечника, характер и количество пищевой массы, особенности регионарного кровотока, состояние соединительной ткани, агрегантное состояние веществ, сочетание лекарственных средств.
Основные фармакокинетические параметры

Фармакокинетика — раздел клинической фармакологии, изучающий пути введения, биотрансформацию, связь с белками крови, распределение и выведение лекарственных средств (ЛС).
Один из основных показателей, определяющих фармакологический эффект, — концентрация ЛС в области рецептора, однако в условиях целостного организма установить её невозможно. Экспериментально доказано, что в большинстве случаев имеется корреляция между концентрацией препарата в крови и его содержанием в других биологических жидкостях и тканях.
Поэтому для определения фармакокинетических параметров ЛС изучают его содержание в крови. Чтобы получить соответствующие представления о поступлении препарата в кровь и выведении его из организма, определяют содержание ЛС в плазме крови в течение длительного времени, используя методы жидкостной или газожидкостной хроматографии, радиоиммунный и иммуноферментный анализы, спектрофотометрический метод. На основании полученных данных строят график (фармакокинетическую кривую), отмечая на оси абсцисс время исследования, а на оси ординат — концентрацию ЛС в плазме крови.
В связи со сложностью описания деталей процесса распределения ЛС во всех органах и тканях, организм условно представляют в виде одной или нескольких изолированных проницаемой мембраной частей (камер), в которых Л С распределяется. Этот вид моделирования называют камерным. За центральную камеру обычно принимают кровь и хорошо кровоснабжаемые органы (сердце, лёгкие, печень, почки, эндокринные железы), за периферическую — менее интенсивно кровоснабжаемые органы и ткани (мышцы, кожу, жировую ткань). В этих камерах ЛС распределяется с разной скоростью: быстрее — в центральной, медленнее — в периферической. К наиболее простым относят однокамерную модель, когда предполагают, что после введения препарата его концентрация убывает по моноэкспоненциальному закону. В соответствии с законами линейной кинетики скорость изменения количества препарата в камере пропорциональна его количеству в этой камере.
Кажущийся объём распределения (Vd) — гипотетический объём жидкости организма, необходимый для равномерного распределения всего количества ЛС (введённой дозы) в концентрации, аналогичной таковой в плазме крови. Этот показатель измеряют в л/кг. При внутривенном введении объём распределения равен отношению дозы ЛС к его начальной концентрации в крови.
• Высокие значения объёма распределения свидетельствуют о том, что ЛС активно проникает в биологические жидкости и ткани. При этом, если ЛС активно связывается, например, жировой тканью, его концентрация в крови может практически мгновенно стать очень низкой, а объём распределения достигнет нескольких сотен литров, превысив реальный объём жидкостей организма. Поэтому этот показатель и называют кажущимся объёмом распределения.
• Объём распределения зависит от различных факторов.
— Физико-химические свойства ЛС (молекулярная масса, степень ионизации и полярности, растворимость в воде и жирах) влияют на его прохождение через мембраны.
— Физиологические факторы (возраст, пол, общее количество жировой ткани в организме). Например, у пожилых людей и ново рождённых Vd снижен.
— Патологические состояния, особенно заболевания печени, почек, сердечно-сосудистой системы (ССС).
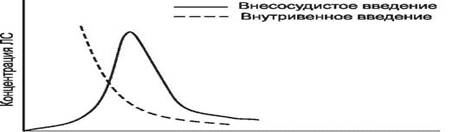
Максимальная концентрация (Сmax) и время наступления максимальной концентрации (Тmax). При поступлении ЛС в системный кровоток (в случае внесосудистого введения) его концентрация постепенно возрастает, достигая значения (Сmax) в момент Тmax, а затем начинает снижаться.
• Если процесс абсорбции имеет линейный характер (скорость процесса прямо пропорциональна количеству ЛС в системе), скорость этого процесса характеризуется константой абсорбции (kabs), измеряемой в часах и рассчитывается через период полувсасывания (Т1/2abs) — время, в течение которого всасывается 1/2 введённой дозы препарата.
Биодоступность (F) — часть дозы Л С (в %), достигшая системного кровотока после вне-сосудистого введения (в этом случае не всё количество препарата достигает системного кровотока).
• Абсолютную биодоступность определяют соотношением значений площади под кинетической кривой (area under curve, AUC) при вне-сосудистом и внутривенном введениях препарата.
— В рамках однокамерной модели при внутривенном введении площадь под кинетической кривой определяется отношением начальной концентрации в крови (Со) к константе элиминации (кеl)
— AUC прямо пропорциональна однократной дозе ЛС, введённой внутривенно (в/в), и обратно пропорциональна общему клиренсу препарата. Она связана с величиной объёма распределения:
где Vd — объём распределения, кеl — константа элиминации, D — доза, AUC — площадь под кинетической кривой.
• Биоэквивалентность (относительная биодоступность) — соотношение количества ЛС, поступившего в системное кровообращение при применении его в различных лекарственных формах или лекарственных препаратах, выпускаемых различными фирмами. Если сравниваемые ЛС аналогичны (действующее вещество, доза, лекарственная t форма), но изготовлены разными производителями, их называют дженериками, и в этом случае необходимо исследование их биоэкви— валентности. Два лекарственных препарата биоэквивалентны, если они обеспечивают одинаковую биодоступность ЛС.
Константа скорости элиминации (кеl) — процент снижения концентрации вещества в крови в единицу времени (отражает долю препарата, выводимую из организма за единицу времени). Элиминация складывается из процессов биотрансформации и экскреции. Константа скорости элиминации характеризует элиминацию в рамках однокамерной модели при линейном характере процесса выведения. Период полувыведения (Т1/2) — время, необходимое для снижения концентрации препарата в крови на 50% в результате элиминации. В рамках линейной модели Т1/2 рассчитывают по формуле:
• Практически за один Т 1 / 2 из организма выводится 50% ЛС, за два периода — 75%, за 3 периода — приблизительно 90% и т.д.
• Зависимость между Т1/2 и кеl важна для подбора режима дозирования и особенно для определения интервала между дозами.
Клиренс (CI) — объём плазмы или крови, полностью освобождающийся от ЛС в единицу времени. Этот показатель количественно характеризует выведение препарата и выражается в мл/мин или л/ч. В рамках линейной модели клиренс рассчитывают по формуле:
• Общий клиренс представляет собой сумму почечного и печёночного клиренсов (так как эти органы служат основными путями выведения ЛС). (Другие пути выведения или внепечёночный метаболизм при расчёте общего клиренса обычно не учитывают.)
— Печёночный клиренс характеризует биотрансформацию ЛС в печени (метаболический клиренс) и выведение с жёлчью (жёлчный клиренс).
— Почечный клиренс отражает выведение препарата с мочой. На пример, почечный клиренс циметидина приблизительно составляет 600 мл/мин, метаболический — 200 мл/мин, жёлчный — 10 мл/мин, поэтому общий клиренс равен 810 мл/мин.
• Основные физиологические факторы, определяющие клиренс, — функциональное состояние основных физиологических систем организма, объём притекающей крови и скорость кровотока в органе. Печёночный клиренс зависит от скорости печёночного кровотока или функциональной способности метаболизирующих ферментов. Например, клиренс лидокаина, интенсивно метаболизируемого печёночными ферментами, зависит прежде всего от скорости его доставки к печени (т.е. от объёма притекающей крови и скорости кровотока), поэтому, например, при застойной сердечной недостаточности он снижен. Клиренс же фенотиазинов зависит в основном от активности метаболизирующих ферментов, поэтому при поражении гепатоцитов клиренс препаратов этой группы резко снижается, вследствие чего концентрация их в крови значительно возрастает.
Равновесная (или стационарная) концентрация (Css) — концентрация, достигнутая при состоянии, когда в каждом интервале между приёмом очередных доз количество всасывающегося ЛС равно количеству элиминируемого [т.е. при стационарном (steady state), или равновесном, состоянии]. Т.е. если ЛС вводят в постоянной дозе через фиксированные интервалы времени, продолжительность которых меньше времени элиминации, его концентрация в крови возрастает, а затем колеблется в пределах средней величины между максимальными и минимальными значениями.
• При достижении С проявляется в полном объёме клинический эффект ЛС. Чем меньше Т1/2 ЛС, тем скорее достигается Си и тем выражение будут её колебания. Например, Т1/2 новокаинамида равен 2— 3 ч, и при назначении через каждые 6 ч его Css характеризуется большим разбросом значений. Поэтому для предупреждения и уменьшения колебаний Css в крови всё большее распространение получают лекарственные формы с замедленным высвобождением активного вещества.
В клинической практике фармакокинетические параметры используют, в частности, для расчёта назначаемых доз препаратов.
• Для расчёта нагрузочной дозы, требуемой для достижения необходимой эффективной концентрации ЛС в крови, используют объём распределения:
где Dнагр — нагрузочная доза, VD — объём распределения, С — концентрация ЛС в плазме крови.
• Для расчёта поддерживающей дозы, т.е. дозы, необходимой для поддержания нужной концентрации ЛС в крови, используют значение клиренса:
где Dnoд — поддерживающая доза, Сl — общий клиренс, Сss — равновесная концентрация.
К основным фармакокинетическим процессам относят всасывание, метаболизм (биотрансформацию), распределение и выведение ЛС.
Публикации в СМИ
Место амлодипина в кардиологической практике
Антагонисты кальция (АК) используются в клинической практике около четырех десятков лет, и в настоящее время это один из самых часто назначаемых препаратов в кардиологии. Такое широкое применение АК в клинической практике связано с их высокой антигипертензивной эффективностью, метаболической нейтральностью и хорошей переносимостью [1, 14]. На российском фармацевтическом рынке в последние годы присутствуют антагонисты кальция дигидропиридинового, фенилалкиламинового и бензотиазепинового рядов, однако наиболее широкое применение в клинической практике нашли производные дигидропиридина: нифедипин, амлодипин, исрадипин, нитрендипин, лацидипин, фелодипин, лерканидипин.
На сегодняшний день самым назначаемым представителем антагонистов кальция дигидропиридинового ряда по праву является амлодипин (Амловас, Амлотоп, Веро-Амлодипин, Калчек, Кардилопин, Корди Кор, Норваск, Нормодипин, Омелар Кардио, Стамло, Тенокс).
Амлодипин — антагонист кальциевых каналов дигидропиридинового ряда третьего поколения, блокирует медленные кальциевые каналы (каналы L-типа) и препятствует внутриклеточной гиперкальциемии и сокращению гладкомышечной клетки, оказывая сосудорасширяющее действие. Обладает длительным действием, что позволяет применять его один раз в сутки.
Фармакокинетика
При приеме внутрь амлодипин медленно и практически полностью всасывается из желудочно-кишечного тракта вне зависимости от приема пищи. Биодоступность амлодипина высока и составляет от 60 до 80%. Объем распределения препарата равен в среднем 20–21 л/кг массы тела, что значительно больше, чем у других представителей дигидропиридинового ряда. В сыворотке 95–98% дозы препарата связывается с белками плазмы. Максимальная концентрация в крови достигается через 6–12 ч после приема. Длительность действия обусловлена его медленным высвобождением из связи с рецепторами. Биотрансформация до неактивных метаболитов происходит в печени. Выводится препарат с мочой (около 10% в неизменном виде и около 60% в виде неактивных метаболитов) и с фекалиями. Период полувыведения равен 35–50 ч. Стабильная равновесная концентрация (steady-state) достигается через 7–8 дней приема препарата. При нарушенной функции печени время выведения амлодипина увеличивается, что характерно и для других антагонистов кальция дигидропиридинового ряда. Амлодипин не вызывает нарушения толерантности к глюкозе и может применяться у больных сахарным диабетом.
Фармакодинамика
Влияние на артериальное давление (АД) и частоту сокращений сердца (ЧСС)
Влияние амлодипина (в виде блокирования медленных кальциевых каналов и снижения внутриклеточной гиперкальциемии) в 80 раз более выражено в отношении гладкомышечных клеток сосудов в сравнении с сократительным миокардом. Таким образом, снижение АД под действием амлодипина происходит именно вследствие периферической вазодилатации. Амлодипин обладает выраженным гипотензивным действием в отношении как систолического (САД), так и диастолического артериального давления (ДАД).
Амлодипин обладает длительным гипотензивным действием за счет большого периода полувыведения (35–50 ч), что позволяет ему контролировать АД равномерно в течение суток, в том числе предупреждает ранние утренние подъемы АД вне зависимости от времени приема препарата (утром или вечером один раз в сутки) [8, 19]. В исследовании Hayduk К. et al. при перерыве в лечении АД сохранялось в пределах нормальных цифр даже на вторые сутки отмены препарата [11]. Максимальный гипотензивный эффект при терапии 5 мг амлодипина наступает лишь на 6-й неделе применения препарата, что делает нецелесообразным раннее увеличение дозы при неполном контроле уровня АД [30]. Препарат оказывает дозозависимое действие на уровень АД и характеризуется линейной зависимостью «доза–концентрация» в плазме крови. Так, при исследовании на здоровых волонтерах ДАД снижалось при измерении стоя на 1,1; 4,8 и 8,0 мм рт. ст., при применении 2,5; 5 и 10 мг амлодипина соответственно [17].
При развитии гипотензивного действия амлодипина не происходит изменения ЧСС, что выделяет препарат среди остальных представителей дигидропиридинового ряда. Кроме того, препарат хорошо переносится пациентами. Среди побочных эффектов — отеки голеней и гиперемия, что свойственно всем АК дигидропиридинового ряда.
Влияние на ишемию миокарда
Антиангинальный эффект определяется особенностью механизма действия и обусловлен коронаролитическим воздействием препарата, что также определяет наиболее предпочтительный контингент больных. Эффект препарата максимален именно у пациентов с выраженным ангиоспазмом [21]. Однако амлодипин также широко применяется для лечения стабильной стенокардии напряжения, достоверно снижая частоту, продолжительность и выраженность эпизодов ишемии миокарда [16, 27]. Одним из возможных благоприятных протективных эффектов амлодипина на состояние миокарда после эпизода ишемии считается его способность снижать кальциевую перегрузку клеток, являющуюся причиной миокардиального повреждения [10]. Выгодным отличием амлодипина от более ранних антагонистов кальция считается отсутствие у него влияния на ЧСС, увеличение которой при физической нагрузке является одним из пусковых механизмов ишемии.
В исследовании CAPE [3] изучены антиишемические свойства амлодипина у больных ИБС. При контрольном 48-часовом мониторировании ЭКГ наблюдалось уменьшение количества эпизодов ишемии миокарда. При добавлении амлодипина к традиционной терапии b-блокаторами и нитратами не наблюдалось возрастания эпизодов нарушений ритма.
Влияние на симпатическую вегетативную нервную систему и активность ренина плазмы
Активация симпатического компонента вегетативной нервной системы является нежелательным побочным эффектом антагонистов кальция дигидропиридинового ряда, обусловленным прямым механизмом их действия. Однако особенностью действия амлодипина является отсутствие активации симпатической нервной системы и развития рефлекторной тахикардии. Так, при определении норадреналина в крови пациентов, принимавших амлодипин, не отмечено повышения его уровня по сравнению с исходным [23]. При спектральном анализе не наблюдалось увеличения показателя LF/HF [24]. Отсутствие влияния амлодипина на активность ренина плазмы и уровень норадреналина у пациентов с артериальной гипертензией (АГ) показано в исследовании Susaguri et al. [24].
Влияние на массу миокарда левого желудочка
Кардиопротективный эффект и способность амлодипина снижать массу миокарда левого желудочка продемонстрированы в рандомизированном, двойном слепом, плацебо-контролируемом исследовании TOMHS, в котором проводилось сравнение 5 антигипертензивных препаратов: диуретика (хлорталидон), β-адреноблокатора (ацебутолол), антагониста кальциевых каналов (амлодипин), ингибитора АПФ (эналаприл), антагониста a-адренорецепторов (доксазозин). При исследовании динамики изменения массы миокарда левого желудочка оказалось, что наиболее выраженное снижение наблюдалось в группах амлодипина и хлорталидона, по сравнению с группами ацебутолола и плацебо, и привело к снижению риска развития сердечно-сосудистых осложнений у больных АГ с гипертрофией левого желудочка.
Влияние на прогрессирование атеросклероза сосудов
Благоприятное влияние АК на состояние эндотелия сосудов подтверждено в многоцентровом, проспективном, рандомизированном, двойном слепом, плацебо-контролируемом исследовании PREVENT [22], в ходе которого оценивалось изменение степени атеросклеротического поражения коронарных артерий и толщины интимомедиального слоя сонных артерий на фоне назначения амлодипина. Отмечалось выраженное влияние амлодипина на прогрессирование атеросклероза в сонных артериях, выявляемое с помощью ультрасонографии. При этом в группе амлодипина наблюдалась регрессия интимомедиального слоя на 0,046 мм, а в группе контроля — утолщение на 0,011 мм. В настоящее время доказана корреляция степени утолщения интимомедиального слоя сонных артерий с частотой развития инфаркта миокарда (ИМ) и мозгового инсульта.
Опыт клинического применения
Артериальная гипертония
Амлодипин эффективно снижает уровни САД и ДАД как при монотерапии, так и в составе комбинированной терапии. Амлодипин является конкурентноспособным препаратом при сравнении с другими антагонистами кальция и препаратами из других классов антигипертензивных средств [9].
При сопоставлении влияния амлодипина с влиянием других антагонистов кальция показана его большая эффективность в отношении уровня АД в сравнении с верапамилом и дилтиаземом. Так, в исследовании Watts R. W. et al. [29] сравнивали амлодипин (5–10 мг) и дилтиазем с контролируемым высвобождением препарата (180–360 мг), при этом среднесуточное АД снижалось при применении амлодипина до 137/84 мм рт. ст., а при применении дилтиазема — до 143/86 мм рт. ст. В многочисленных сравнениях амлодипина с другими дигидропиридиновыми антагонистами кальция он оказывал сопоставимое по величине, но значительно более длительное антигипертензивное действие [8, 13]. При этом выгодным преимуществом препарата перед другими антагонистами кальция дигидропиридинового ряда является отсутствие его влияния на ЧСС, что позволяет назначать его пациентам с тахисистолией.
С точки зрения снижения риска развития сердечно-сосудистых осложнений и улучшения прогноза при АГ (основная цель в лечении этого заболевания), амлодипин, по данным таких исследований, как ALLHAT [25] и VALUE [12], оказался сопоставим по эффективности с ингибиторами АПФ и блокаторами ангиотензиновых рецепторов, а по некоторым позициям даже лучше. В сравнении с валсартаном режим антигипертензивной терапии на основе амлодипина достоверно снизил частоту развития ИМ на 19% у больных АГ с многочисленными сопутствующими факторами риска.
Ангиопротективные эффекты амлодипина продемонстрированы в исследовании PREVENT [22]. Было показано, что у больных ишемической болезнью сердца (ИБС) амлодипин способствовал уменьшению величины показателя «толщина интима-медиа» по сравнению с группой больных ИБС, получавших плацебо. Возможно, именно замедлением прогрессирования атеросклероза сонных артерий объясняются данные, полученные во многих исследованиях, указывающих на особенно выраженное снижение частоты развития нарушений мозгового кровообращения при лечении антагонистами кальция [28]. Результаты исследования CAMELOT [18] позволяют судить об антиатеросклеротическом действии амлодипина и в отношении коронарных артерий.
Важнейшим звеном органопротекции при терапии больных АГ является профилактика инсульта. В исследовании ASCOT сравнивалась эффективность гипотензивной терапии, основанной на применении антагониста кальция амлодипина с последующим присоединением ингибитора АПФ периндоприла, с терапией, основанной на применении b-блокатора атенолола и тиазидного диуретика бендрофлюметиазида. В обеих группах наблюдалось сопоставимое снижение уровня АД, однако в группе лечения амлодипином и ингибитором АПФ относительный риск развития инсульта был на 23% ниже, чем в группе лечения атенололом и диуретиком (p = 0,0003) [4]. Результаты исследования ASCOT позволяют предположить, что, помимо гипотензивного действия, антагонисты кальция и ингибиторы АПФ обладают дополнительными свойствами, позволяющими снизить риск развития цереброваскулярных осложнений.
Важнейшей характеристикой антигипертензивного лекарственного средства являются его метаболические эффекты, в частности, влияние на углеводный обмен [26]. Поскольку развитие СД у больных АГ значительно увеличивает риск сердечно-сосудистых осложнений [7], подбор гипотензивной терапии необходимо проводить с учетом ее влияния на риск возникновения СД.Антагонисты кальция уменьшают частоту возникновения новых случаев СД по сравнению с терапией диуретиками [12, 25]. В исследовании ALLHAT, в зависимости от частоты новых случаев СД в группах сравнения, тестируемые препараты распределились следующим образом: хлорталидон > амлодипин > лизиноприл [25]. Однако особенно ярко благоприятное влияние амлодипина проявилось в исследовании ASCOT [4], где на фоне лечения амлодипином в комбинации с периндоприлом вероятность развития новых случаев СД была на 30% меньше, чем в группе больных, получавших терапию атенололом и тиазидовым диуретиком бендрофлюметиазидом.
Результаты проведенных крупных исследований (PREVENT, INSIGHT, ELSA, CAMELOT и др.) послужили предпосылкой для расширения показаний к использованию АК у больных АГ и внесения в новые рекомендации наличия атеросклероза сонных и коронарных артерий у больных АГ в качестве одного из показаний для первоочередного назначения АК дигидропиридиновой группы [26]. Кроме того, в Европейских рекомендациях 2007 г. антагонисты кальция из группы дигидропиридиновых производных (амлодипин) показаны как препараты выбора у пожилых пациентов с изолированной систолической АГ, стенокардией, гипертрофией миокарда левого желудочка, заболеваниями периферических сосудов, при беременности, атеросклерозе сонных и коронарных артерий.
Ишемическая болезнь сердца. Стенокардия
Амлодипин обладает выраженным коронаролитическим эффектом вследствие высокой селективности в отношении гладкомышечных клеток артериол. Возникающий при применении короткодействующих АК синдром обкрадывания значительно менее выражен у препаратов длительного действия, в частности у амлодипина. Амлодипин широко применяется для терапии стабильной стенокардии напряжения как в монотерапии, так (чаще) и в составе комбинированной терапии. Благоприятный эффект амлодипин оказывает и при применении у пациентов с выраженным динамическим компонентом коронарной обструкции [3].
Антиишемические эффекты амлодипина, его способность снижать частоту эпизодов депрессии сегмента ST, общее время ишемии (по данным ЭКГ), а также частоту болевых эпизодов ишемии и кратность дополнительного применения короткодействующих нитратов, продемонстрированы в ряде исследований, в том числе в многоцентровом исследовании CAPE [5, 6].
Влияние амлодипина на прогноз у пациентов с ИБС было оценено в исследовании PREVENT [22]. Наблюдалось уменьшение числа госпитализаций, обусловленных дестабилизацией течения стенокардии и хронической сердечной недостаточности (ХСН); уменьшение числа операций реваскуляризации миокарда (53 в сравнении с 85 в группе плацебо) вне зависимости от применения β-блокаторов, нитратов или липидснижающей терапии.
В метаанализе Kloner R. A. et al. [15] оценивалась безопасность применения антагонистов кальциевых каналов. Были включены сравнительные и несравнительные исследования амлодипина и нифедипина GITS. Показано, что у пациентов, получавших амлодипин, общая сердечно-сосудистая летальность, частота развития острого ИМ и прогрессирования ИБС была значительно ниже аналогичных показателей для других антагонистов кальция.
По данным исследования CAMELOT амлодипин по сравнению с плацебо на 31% (р
Код вставки на сайт
Место амлодипина в кардиологической практике
Антагонисты кальция (АК) используются в клинической практике около четырех десятков лет, и в настоящее время это один из самых часто назначаемых препаратов в кардиологии. Такое широкое применение АК в клинической практике связано с их высокой антигипертензивной эффективностью, метаболической нейтральностью и хорошей переносимостью [1, 14]. На российском фармацевтическом рынке в последние годы присутствуют антагонисты кальция дигидропиридинового, фенилалкиламинового и бензотиазепинового рядов, однако наиболее широкое применение в клинической практике нашли производные дигидропиридина: нифедипин, амлодипин, исрадипин, нитрендипин, лацидипин, фелодипин, лерканидипин.
На сегодняшний день самым назначаемым представителем антагонистов кальция дигидропиридинового ряда по праву является амлодипин (Амловас, Амлотоп, Веро-Амлодипин, Калчек, Кардилопин, Корди Кор, Норваск, Нормодипин, Омелар Кардио, Стамло, Тенокс).
Амлодипин — антагонист кальциевых каналов дигидропиридинового ряда третьего поколения, блокирует медленные кальциевые каналы (каналы L-типа) и препятствует внутриклеточной гиперкальциемии и сокращению гладкомышечной клетки, оказывая сосудорасширяющее действие. Обладает длительным действием, что позволяет применять его один раз в сутки.
Фармакокинетика
При приеме внутрь амлодипин медленно и практически полностью всасывается из желудочно-кишечного тракта вне зависимости от приема пищи. Биодоступность амлодипина высока и составляет от 60 до 80%. Объем распределения препарата равен в среднем 20–21 л/кг массы тела, что значительно больше, чем у других представителей дигидропиридинового ряда. В сыворотке 95–98% дозы препарата связывается с белками плазмы. Максимальная концентрация в крови достигается через 6–12 ч после приема. Длительность действия обусловлена его медленным высвобождением из связи с рецепторами. Биотрансформация до неактивных метаболитов происходит в печени. Выводится препарат с мочой (около 10% в неизменном виде и около 60% в виде неактивных метаболитов) и с фекалиями. Период полувыведения равен 35–50 ч. Стабильная равновесная концентрация (steady-state) достигается через 7–8 дней приема препарата. При нарушенной функции печени время выведения амлодипина увеличивается, что характерно и для других антагонистов кальция дигидропиридинового ряда. Амлодипин не вызывает нарушения толерантности к глюкозе и может применяться у больных сахарным диабетом.
Фармакодинамика
Влияние на артериальное давление (АД) и частоту сокращений сердца (ЧСС)
Влияние амлодипина (в виде блокирования медленных кальциевых каналов и снижения внутриклеточной гиперкальциемии) в 80 раз более выражено в отношении гладкомышечных клеток сосудов в сравнении с сократительным миокардом. Таким образом, снижение АД под действием амлодипина происходит именно вследствие периферической вазодилатации. Амлодипин обладает выраженным гипотензивным действием в отношении как систолического (САД), так и диастолического артериального давления (ДАД).
Амлодипин обладает длительным гипотензивным действием за счет большого периода полувыведения (35–50 ч), что позволяет ему контролировать АД равномерно в течение суток, в том числе предупреждает ранние утренние подъемы АД вне зависимости от времени приема препарата (утром или вечером один раз в сутки) [8, 19]. В исследовании Hayduk К. et al. при перерыве в лечении АД сохранялось в пределах нормальных цифр даже на вторые сутки отмены препарата [11]. Максимальный гипотензивный эффект при терапии 5 мг амлодипина наступает лишь на 6-й неделе применения препарата, что делает нецелесообразным раннее увеличение дозы при неполном контроле уровня АД [30]. Препарат оказывает дозозависимое действие на уровень АД и характеризуется линейной зависимостью «доза–концентрация» в плазме крови. Так, при исследовании на здоровых волонтерах ДАД снижалось при измерении стоя на 1,1; 4,8 и 8,0 мм рт. ст., при применении 2,5; 5 и 10 мг амлодипина соответственно [17].
При развитии гипотензивного действия амлодипина не происходит изменения ЧСС, что выделяет препарат среди остальных представителей дигидропиридинового ряда. Кроме того, препарат хорошо переносится пациентами. Среди побочных эффектов — отеки голеней и гиперемия, что свойственно всем АК дигидропиридинового ряда.
Влияние на ишемию миокарда
Антиангинальный эффект определяется особенностью механизма действия и обусловлен коронаролитическим воздействием препарата, что также определяет наиболее предпочтительный контингент больных. Эффект препарата максимален именно у пациентов с выраженным ангиоспазмом [21]. Однако амлодипин также широко применяется для лечения стабильной стенокардии напряжения, достоверно снижая частоту, продолжительность и выраженность эпизодов ишемии миокарда [16, 27]. Одним из возможных благоприятных протективных эффектов амлодипина на состояние миокарда после эпизода ишемии считается его способность снижать кальциевую перегрузку клеток, являющуюся причиной миокардиального повреждения [10]. Выгодным отличием амлодипина от более ранних антагонистов кальция считается отсутствие у него влияния на ЧСС, увеличение которой при физической нагрузке является одним из пусковых механизмов ишемии.
В исследовании CAPE [3] изучены антиишемические свойства амлодипина у больных ИБС. При контрольном 48-часовом мониторировании ЭКГ наблюдалось уменьшение количества эпизодов ишемии миокарда. При добавлении амлодипина к традиционной терапии b-блокаторами и нитратами не наблюдалось возрастания эпизодов нарушений ритма.
Влияние на симпатическую вегетативную нервную систему и активность ренина плазмы
Активация симпатического компонента вегетативной нервной системы является нежелательным побочным эффектом антагонистов кальция дигидропиридинового ряда, обусловленным прямым механизмом их действия. Однако особенностью действия амлодипина является отсутствие активации симпатической нервной системы и развития рефлекторной тахикардии. Так, при определении норадреналина в крови пациентов, принимавших амлодипин, не отмечено повышения его уровня по сравнению с исходным [23]. При спектральном анализе не наблюдалось увеличения показателя LF/HF [24]. Отсутствие влияния амлодипина на активность ренина плазмы и уровень норадреналина у пациентов с артериальной гипертензией (АГ) показано в исследовании Susaguri et al. [24].
Влияние на массу миокарда левого желудочка
Кардиопротективный эффект и способность амлодипина снижать массу миокарда левого желудочка продемонстрированы в рандомизированном, двойном слепом, плацебо-контролируемом исследовании TOMHS, в котором проводилось сравнение 5 антигипертензивных препаратов: диуретика (хлорталидон), β-адреноблокатора (ацебутолол), антагониста кальциевых каналов (амлодипин), ингибитора АПФ (эналаприл), антагониста a-адренорецепторов (доксазозин). При исследовании динамики изменения массы миокарда левого желудочка оказалось, что наиболее выраженное снижение наблюдалось в группах амлодипина и хлорталидона, по сравнению с группами ацебутолола и плацебо, и привело к снижению риска развития сердечно-сосудистых осложнений у больных АГ с гипертрофией левого желудочка.
Влияние на прогрессирование атеросклероза сосудов
Благоприятное влияние АК на состояние эндотелия сосудов подтверждено в многоцентровом, проспективном, рандомизированном, двойном слепом, плацебо-контролируемом исследовании PREVENT [22], в ходе которого оценивалось изменение степени атеросклеротического поражения коронарных артерий и толщины интимомедиального слоя сонных артерий на фоне назначения амлодипина. Отмечалось выраженное влияние амлодипина на прогрессирование атеросклероза в сонных артериях, выявляемое с помощью ультрасонографии. При этом в группе амлодипина наблюдалась регрессия интимомедиального слоя на 0,046 мм, а в группе контроля — утолщение на 0,011 мм. В настоящее время доказана корреляция степени утолщения интимомедиального слоя сонных артерий с частотой развития инфаркта миокарда (ИМ) и мозгового инсульта.
Опыт клинического применения
Артериальная гипертония
Амлодипин эффективно снижает уровни САД и ДАД как при монотерапии, так и в составе комбинированной терапии. Амлодипин является конкурентноспособным препаратом при сравнении с другими антагонистами кальция и препаратами из других классов антигипертензивных средств [9].
При сопоставлении влияния амлодипина с влиянием других антагонистов кальция показана его большая эффективность в отношении уровня АД в сравнении с верапамилом и дилтиаземом. Так, в исследовании Watts R. W. et al. [29] сравнивали амлодипин (5–10 мг) и дилтиазем с контролируемым высвобождением препарата (180–360 мг), при этом среднесуточное АД снижалось при применении амлодипина до 137/84 мм рт. ст., а при применении дилтиазема — до 143/86 мм рт. ст. В многочисленных сравнениях амлодипина с другими дигидропиридиновыми антагонистами кальция он оказывал сопоставимое по величине, но значительно более длительное антигипертензивное действие [8, 13]. При этом выгодным преимуществом препарата перед другими антагонистами кальция дигидропиридинового ряда является отсутствие его влияния на ЧСС, что позволяет назначать его пациентам с тахисистолией.
С точки зрения снижения риска развития сердечно-сосудистых осложнений и улучшения прогноза при АГ (основная цель в лечении этого заболевания), амлодипин, по данным таких исследований, как ALLHAT [25] и VALUE [12], оказался сопоставим по эффективности с ингибиторами АПФ и блокаторами ангиотензиновых рецепторов, а по некоторым позициям даже лучше. В сравнении с валсартаном режим антигипертензивной терапии на основе амлодипина достоверно снизил частоту развития ИМ на 19% у больных АГ с многочисленными сопутствующими факторами риска.
Ангиопротективные эффекты амлодипина продемонстрированы в исследовании PREVENT [22]. Было показано, что у больных ишемической болезнью сердца (ИБС) амлодипин способствовал уменьшению величины показателя «толщина интима-медиа» по сравнению с группой больных ИБС, получавших плацебо. Возможно, именно замедлением прогрессирования атеросклероза сонных артерий объясняются данные, полученные во многих исследованиях, указывающих на особенно выраженное снижение частоты развития нарушений мозгового кровообращения при лечении антагонистами кальция [28]. Результаты исследования CAMELOT [18] позволяют судить об антиатеросклеротическом действии амлодипина и в отношении коронарных артерий.
Важнейшим звеном органопротекции при терапии больных АГ является профилактика инсульта. В исследовании ASCOT сравнивалась эффективность гипотензивной терапии, основанной на применении антагониста кальция амлодипина с последующим присоединением ингибитора АПФ периндоприла, с терапией, основанной на применении b-блокатора атенолола и тиазидного диуретика бендрофлюметиазида. В обеих группах наблюдалось сопоставимое снижение уровня АД, однако в группе лечения амлодипином и ингибитором АПФ относительный риск развития инсульта был на 23% ниже, чем в группе лечения атенололом и диуретиком (p = 0,0003) [4]. Результаты исследования ASCOT позволяют предположить, что, помимо гипотензивного действия, антагонисты кальция и ингибиторы АПФ обладают дополнительными свойствами, позволяющими снизить риск развития цереброваскулярных осложнений.
Важнейшей характеристикой антигипертензивного лекарственного средства являются его метаболические эффекты, в частности, влияние на углеводный обмен [26]. Поскольку развитие СД у больных АГ значительно увеличивает риск сердечно-сосудистых осложнений [7], подбор гипотензивной терапии необходимо проводить с учетом ее влияния на риск возникновения СД.Антагонисты кальция уменьшают частоту возникновения новых случаев СД по сравнению с терапией диуретиками [12, 25]. В исследовании ALLHAT, в зависимости от частоты новых случаев СД в группах сравнения, тестируемые препараты распределились следующим образом: хлорталидон > амлодипин > лизиноприл [25]. Однако особенно ярко благоприятное влияние амлодипина проявилось в исследовании ASCOT [4], где на фоне лечения амлодипином в комбинации с периндоприлом вероятность развития новых случаев СД была на 30% меньше, чем в группе больных, получавших терапию атенололом и тиазидовым диуретиком бендрофлюметиазидом.
Результаты проведенных крупных исследований (PREVENT, INSIGHT, ELSA, CAMELOT и др.) послужили предпосылкой для расширения показаний к использованию АК у больных АГ и внесения в новые рекомендации наличия атеросклероза сонных и коронарных артерий у больных АГ в качестве одного из показаний для первоочередного назначения АК дигидропиридиновой группы [26]. Кроме того, в Европейских рекомендациях 2007 г. антагонисты кальция из группы дигидропиридиновых производных (амлодипин) показаны как препараты выбора у пожилых пациентов с изолированной систолической АГ, стенокардией, гипертрофией миокарда левого желудочка, заболеваниями периферических сосудов, при беременности, атеросклерозе сонных и коронарных артерий.
Ишемическая болезнь сердца. Стенокардия
Амлодипин обладает выраженным коронаролитическим эффектом вследствие высокой селективности в отношении гладкомышечных клеток артериол. Возникающий при применении короткодействующих АК синдром обкрадывания значительно менее выражен у препаратов длительного действия, в частности у амлодипина. Амлодипин широко применяется для терапии стабильной стенокардии напряжения как в монотерапии, так (чаще) и в составе комбинированной терапии. Благоприятный эффект амлодипин оказывает и при применении у пациентов с выраженным динамическим компонентом коронарной обструкции [3].
Антиишемические эффекты амлодипина, его способность снижать частоту эпизодов депрессии сегмента ST, общее время ишемии (по данным ЭКГ), а также частоту болевых эпизодов ишемии и кратность дополнительного применения короткодействующих нитратов, продемонстрированы в ряде исследований, в том числе в многоцентровом исследовании CAPE [5, 6].
Влияние амлодипина на прогноз у пациентов с ИБС было оценено в исследовании PREVENT [22]. Наблюдалось уменьшение числа госпитализаций, обусловленных дестабилизацией течения стенокардии и хронической сердечной недостаточности (ХСН); уменьшение числа операций реваскуляризации миокарда (53 в сравнении с 85 в группе плацебо) вне зависимости от применения β-блокаторов, нитратов или липидснижающей терапии.
В метаанализе Kloner R. A. et al. [15] оценивалась безопасность применения антагонистов кальциевых каналов. Были включены сравнительные и несравнительные исследования амлодипина и нифедипина GITS. Показано, что у пациентов, получавших амлодипин, общая сердечно-сосудистая летальность, частота развития острого ИМ и прогрессирования ИБС была значительно ниже аналогичных показателей для других антагонистов кальция.
По данным исследования CAMELOT амлодипин по сравнению с плацебо на 31% (р Copy



