Синдром Лея (болезнь Лея)

Общие сведения
Синдром Лея относится к редким нейрометаболическим синдромам, поражающим ЦНС, и вызывающим нарушение координации движений и мышления и летальный исход. Заболевание наследственное по аутосомно-рецессивному типу или Х-сцепленному. Патологии чаще всего подвержены маленькие детки до 2-х лет – 1-2 ребенка на десятки тысяч, в более редких случаях – подростки и взрослые особы. Код по МКБ-10: G 31.8
Миру стало известно об этом заболевании благодаря Денису Лею, который описал его еще в 1951 году. В научных кругах обособить синдром от подобных энцефалопатий удалось в 1954 г. Связь с митохондриальной активностью смогли выявить 1968 г, но главным продвижением стало обнаружение мутации в цитохромоксидазах в 1977 г.
Проблема состоит в том, что эффективных препаратов или методов лечения болезни Лея до сих пор не разработано, ведь причин её множество, начиная с влияния мутаций в генах, отвечающих за работу митохондрий, и заканчивая – осложнениями дегенеративных заболеваний нервной системы, аномальными образованиями некротизированных очагов или прорастания сосудов, глиоза в структурах головного мозга.
Единственным профилактическим методом на сегодня является «рождение детей от трех родителей», то есть благодаря использованию донорских яйцеклеток и митохондриальной донации.
Патогенез
Для таких митохондриальных заболеваний как синдром Лея характерно полиорганное поражение с вовлечением нервных и мышечных тканей. В основе патологии обычно лежит сбой регулирования обмена пировиноградной кислоты, при этом наблюдается нарушение обменных реакций, снижение выработки энергии и замедление процессов перемещения электронов в клетках дыхательной системы. Процессы могут спорадически усилиться, чему способствует наследственные факторы. Для запуска развития синдрома необходимо присутствие в организме более 90% мутантной мтДНК от всей мтДНК. В случае меньшего содержания мутантной ДНК симптоматика напоминает нейропатию и сводится к атаксии и пигментному ретиниту.
Для болезни характерно раннее развитие и стремительное злокачественное течение с присоединением большого количества различных неврологических нарушений, затем начинаются проблемы с дыханием и метаболизмом.
Классификация
В зависимости от патогенеза и возрастных особенностей выделяют различные варианты нарушений, выражающиеся в виде:
В зависимости от течения синдром Лея бывает:
Причины
Главным провоцирующим фактором синдрома Лея принято считать недостаточность цитохромоксидазы и мутации генов, отвечающих за работу митохондрий:
Однако, способствовать нейрометаболической энцефалопатии может:
Симптомы
Синдром Лея и его прогрессирование приводит:
Клиническая характеристика синдрома Лея при других формах патологии
Клиническая характеристика и патогенез схожи, но симптоматика может дополняться:
Анализы и диагностика
Болезнь Лея относится к подострым либо хроническим некротизирующим энцефаломиопатиям и является важным направлением изучения в детской невралгии. Для её диагностики необходима консультация у врача-невролога, а также:
Лечение
Медикаментозное консервативное лечение болезни Лея обычно симптоматическое и включает использование:
Что такое синдром лея
а) Терминология:
1. Сокращения:
• Синдром Лея (ЛС)
2. Синонимы:
• Подострая некротизирующая энцефаломиелопатия
3. Определение:
• Генетически гетерогенное митохондриальное заболевание, характеризующееся прогрессирующей нейродегенерацией
2. КТ признаки синдрома Лея:
• Бесконтрастная КТ:
о Низкая плотность; иногда нормальная рентгенологическая картина
• КТ с контрастированием
о Накопление контрастного вещества нехарактерно
3. МРТ признаки синдрома Лея:
• Т1-ВИ:
о Гипоинтенсивный сигнал:
— Возможно наличие гиперинтенсивных участков = кровь или некроз
• Т2-ВИ:
о Гиперинтенсивный сигнал
• FLAIR:
о Гиперинтенсивный сигнал:
— В хроническую стадию заболевания может наблюдаться разрешение участков изменения сигнала или кистозной энцефаломаляции (гипоинтенсивный сигнал)
• ДВИ:
о Ограничение диффузии в зоне острого поражения
• МР-спектроскопия:
о ↑ пика холина, ↓ пика NAA
о Часто присутствует пик лактата; может быть высоким
4. УЗИ:
• Гиперэхогенность глубоких ядер, БВ
5. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ с ДВИ/МР-спектроскопия
в) Дифференциальная диагностика синдрома Лея:
2. Митохондриальная энцефалопатия, лактоацидоз, инсультоподобные эпизоды (MELAS):
• ↑ интенсивности сигнала на T2-BИ/FLAIR от скорлупы (кальцификация в хроническую стадию)
о Возможна асимметричность или односторонность поражения
• Инсультоподобные изменения сигнальных характеристик в теменно-затылочных областях полушарий:
о Характерно несоответствие бассейнам кровоснабжения и ДВИ(-)
3. Глутаровая ацидурия 1-го типа (ГА1):
• ↑ интенсивности сигнала на T2-BИ/FLAIR от полосатых тел, БШ ± поражение БВ
• Характерное расширение височных покрышек
4. Болезнь Вильсона-Коновалова:
• ↑ интенсивности сигнала на Т2-ВИ/FLAIR от скорлупы, БШ, средний мозг, таламусы:
о Изменения на Т2-ВИ очевидны у детей более старшего возраста, подростков
• Гиперинтенсивный на Т1-ВИ сигнал от БШ вследствие печеночной недостаточности
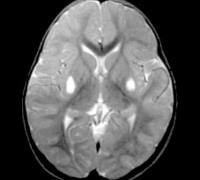
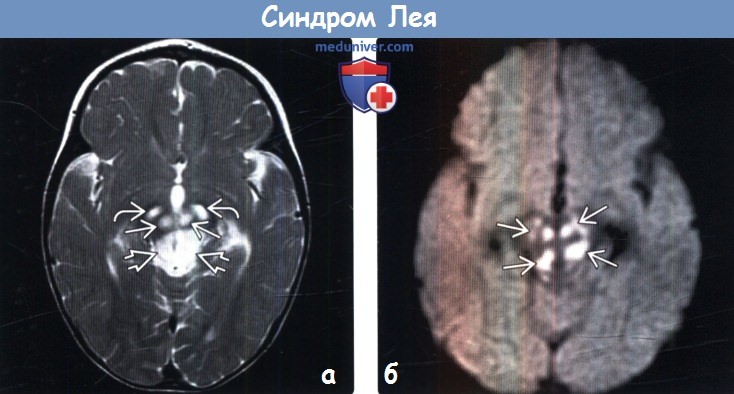 (а) МРТ, Т2-ВИ, аксиальный срез: отмечается повышение интенсивности сигнала от ножек мозга, красных ядер и покрышки среднего мозга (включая периакведуктальное серое вещество). Эти структуры являются частой локализацией поражения ствола мозга при синдроме Лея.
(а) МРТ, Т2-ВИ, аксиальный срез: отмечается повышение интенсивности сигнала от ножек мозга, красных ядер и покрышки среднего мозга (включая периакведуктальное серое вещество). Эти структуры являются частой локализацией поражения ствола мозга при синдроме Лея.
(б) МРТ, ДВИ, аксиальный срез: определяется ограничение диффузии (гиперинтенсивные участки) в пораженных участках среднего мозга. Ограничение диффузии указывает на острую стадию поражения, в то время как повышение диффузии больше указывает на хроническую стадию поражения.
2. Макроскопические и хирургические особенности:
• Коричневато-серые желатинозные или полостные участки в полосатых телах, БШ, СтМ, зубчатых ядрах, таламусах, спинном мозге, белом веществе
3. Микроскопия:
• Спонгиозная дегенерация, глиоз, потеря нейронов, демиелиниза-ция, пролиферация капилляров
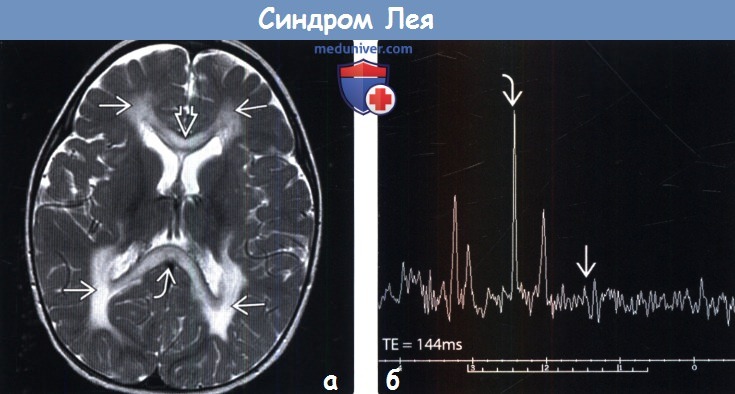 (а) МРТ, Т2-ВИ, аксиальный срез: определяется повышение интенсивности сигнала от колена и валика мозолистого тела, распространяющееся на пери вентрикулярное и глубокое белое вещество, а также задние бедра внутренних капсул.
(а) МРТ, Т2-ВИ, аксиальный срез: определяется повышение интенсивности сигнала от колена и валика мозолистого тела, распространяющееся на пери вентрикулярное и глубокое белое вещество, а также задние бедра внутренних капсул.
(б) Одновоксельная протонная МР-спектроскопия (ТЕ = 144 мс): у того же пациента определяется минимальный пик лактата на 1,33 ppm и аномальный пик на 2,4 ppm, что соответствует сукцинату. Был поставлен диагноз «дефицит сукцинатдегидрогеназы вследствие БСНА-мутации».
д) Клиническая картина:
1. Проявления синдрома Лея:
• Наиболее частые признаки/симптомы:
о Проявления: задержка/регрессия психомоторного развития, гипотония
— Синдром Лея (СЛ) является клиническим диагнозом; вызывать данные проявления могут многие митохондриальные заболевания
о Другие признаки/симптомы:
— Прогрессирующая дисфункция СтМ и БГ:
Атаксия, офтальмоплегия, птоз, рвота, нарушения проглатывания и дыхания, дистония
о Раннее проявление заболевания, дисфункция СтМ, периферическая нейропатия и быстрое неврологическое ухудшение характерно для СЛ, вызванного SURF1-мутацией
о Метаболические стрессовые факторы (например, инфекция) могут «демаскировать» заболевание или вызвать ее ухудшение о Повышение лактата в СМЖ, сыворотке крови, моче является классическим признаком, но наблюдается не всегда
о Клинический диагноз:
— Прогрессирующая нейродегенерация
— Признаки/симптомы дисфункции СтМ и БГ
— ↑ лактата в крови + СМЖ
— Биохимический дефект, выявленный с помощью митохондриального анализа биоптата скелетных мышц или культивируемых фибробластов кожи
— МРТ → характерные поражения БГ или СтМ
о Пренатальная диагностика: проба ворсинчатого хориона (мутации и биохимические дефекты)
• Клинический профиль:
о Младенец с психомоторной регрессией, гипотонией
2. Демография:
• Возраст:
о У большинства заболевание проявляется в возрасте двух лет
о Манифестация заболевания в более старшем детском и взрослом возрасте встречается нечасто
• Половая принадлежность:
о Отсутствует
• Этническая принадлежность:
о Отсутствует
• Эпидемиология:
о Митохондриальные заболевания = 1: 8500
о СЛ у детей
Редактор: Искандер Милевски. Дата публикации: 21.4.2019
Синдром Лея
Причины
Заболевание возникает в результате дефицита ферментов, которые обеспечивают образование энергии, что происходит из-за нарушения обмена пировиноградной кислоты и дефекта транспорта электронов в дыхательной цепи. Причины синдрома Лея:
Симптомы синдрома Лея
Характерные для синдрома Лея симптомы выглядят следующим образом:
Если Вы обнаружили у себя схожие симптомы, незамедлительно обратитесь к врачу. Легче предупредить болезнь, чем бороться с последствиями.
Диагностика
Для диагностики синдрома Лея врач-невролог проводит ряд исследований и анализов, среди которых:
Лечение синдрома Лея
Для лечения синдрома Лея пациентам назначаются:
Диета
В период лечения пациентам назначается диета, согласно которой суточное потребление белка не должно превышать 1–1,5 г/сутки.
Опасность
Отсутствие необходимой помощи при остром течении заболевания приводит к риску возникновения паралича дыхательного центра, прогрессирующей энцефалопатии, сопутствующих инфекций, что приводит к летальному исходу.
Группа риска
В группе риска находятся:
Профилактика
Для профилактики заболевания рекомендуется:
Данная статья размещена исключительно в познавательных целях и не является научным материалом или профессиональным медицинским советом.
Синдром Лея

Синдром Лея (СЛ) — это гетерогенное генетически обусловленное заболевание, относящееся к группе митохондриальных энцефаломиопатий с аутосомно-рецессивным или митохондриальным типом наследования и связанное с мутациями в генах, кодирующих полипептиды комплексов дыхательной цепи митохондрий, а также белков, принимающих участие в их сборке на внутренней поверхности митохондриальной мембраны [4].
Первые описания данного синдрома были даны Денисом Леем в 1951 году [1].
На заре открытия данного заболевания появились предположения, что его причиной является нарушение метаболизма, однако они не получили своего клинического подтверждения.
Доктор F. Hommes в 1968 году описал семьи, у представителей которых наблюдалось снижение активности пируваткарбоксилазы [2]. С 1975 года появились данные, что причиной СЛ может являться недостаточность пируватдегидрогеназного комплекса, а позже были обнаружены молекулярно-генетические нарушения. С развитием генетики и биохимии было выяснено, что изменения генов в мтДНК, которые ответственны за кодирование субъединицы АТФ-азы или тРНК — ядерных генов, кодирующих полипептиды комплекса дыхательной цепи митохондрий, — а также за нарушения в генах, отвечающих за сборку КДЦМ на митохондриальной мембране, приводят к развитию синдрома Лея [3]. Всего насчитывается 12 генов: NDUFS4, NDUFS5, NDUFS6, NDUFS7, NDUFS8, NDUFV1, SDHA, SURF1, COX10, COX15, SCO2, BCS1L [1].
Чаще всего дефект обнаруживается в результате недостаточности IV КДЦМ-цитохром С-оксидазе (COX). Этот фермент является последним в электронно-транспортной системе митохондрий. СОХ включает в себя 13 субъединиц: 11 кодируются ядерными генами, 3 – мтДНК.
Чаще всего данная патология возникает в результате мутации гена SURF1, который лежит в хромосоме 9q34, кластере 6. Данный белок включен во внутреннюю мембрану митохондрии, и нарушения в нем приводят к синтезу укороченного белка и, как следствие, к повреждению СОХ-комплекса [4].
В результате биохимических исследований у большинства пациентов с СЛ обнаруживается повышение лактата в крови и спинномозговой жидкости. Повышение соотношения лактат/пируват является отражением нарушения окислительно-восстановительного баланса в цитоплазме [4].
Те пациенты, у которых мутации не превышают 70% от всех мтДНК к конкретной ткани, не имеют клинических проявлений данного синдрома. Однако при превышении данного порога могут проявляться симптомы, которые будут описаны ниже [1].
Синдром Лея чаще всего проявляется в детстве в виде утраты уже имеющихся психомоторных навыков, мозжечковыми и экстрапирамидными расстройствами, судорогами, мышечной гипотонией. Однако могут проявляться и такие симптомы как нистагм, потеря слуха, атрофия зрительного нерва. Дети отстают в своем развитии, отмечается регресс психомоторных функций, вялость, сонливость. Такие пациенты склонны к лактоацидозам. Ночью и при физических нагрузках возникают проблемы с дыханием в виде апноэ, диспноэ, тахипноэ [1].
При МРТ-диагностике наблюдается снижение МР-сигнала в различных областях ГМ.
Также пациентам с синдромом Лея свойственна «парадоксальная гиперкетонемия» — повышение уровня кетоновых тел после пищевой нагрузки и высокое соотношение 3-гидроксибутират/ацетоацетат в крови. При проведении анализа органических кислот мочи может наблюдаться повышенная экскреция органических кислот, участвующих в цикле Кребса (фумаровая, янтарная и др.) [4].
Синдром Лея имеет следующую классификацию:
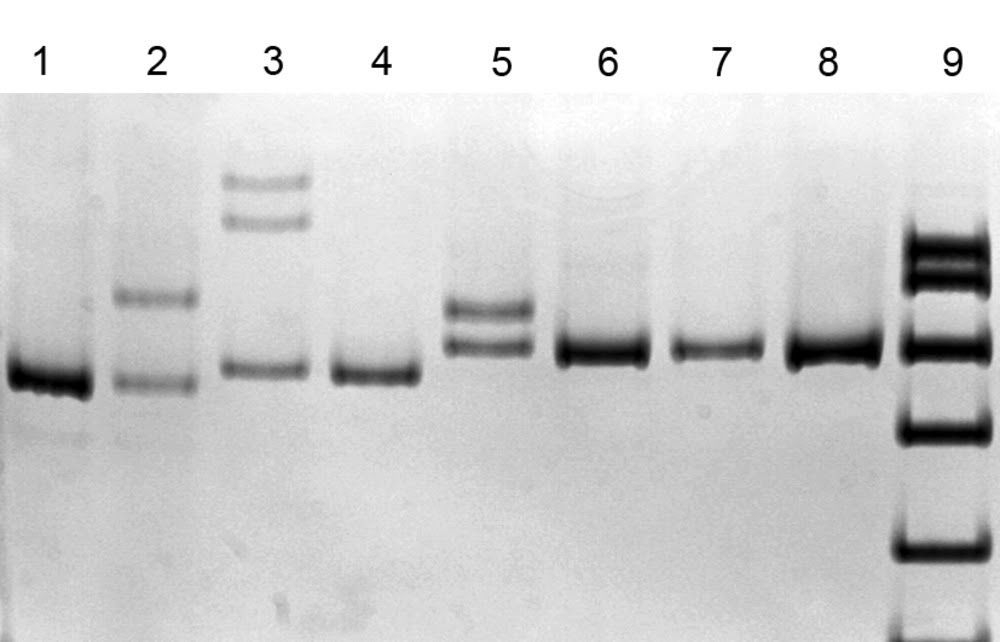
ДНК-диагностика 3 частых мутаций в гене SURF1 с помощью метода SSCP анализа (электрофорез в 8% полиакриламидном геле) [4].
.
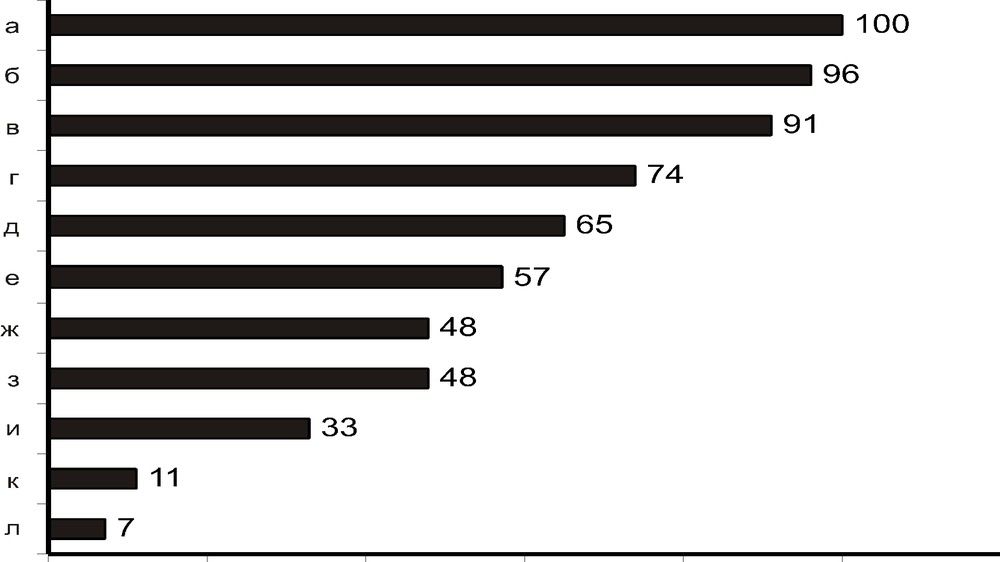
Статистика основных синдромов [4]:
Источники:
Синдром Ли ( Подострая некротизирующая энцефаломиелопатия )
Синдром Ли – генетически гетерогенное наследственное заболевание, характеризующееся разнообразными нарушениями метаболизма и формирования компонентов центральной нервной системы. Симптомы этой патологии, как правило, проявляются еще в раннем детстве, к ним относят мышечную гипотонию, проблемы со вскармливанием и задержку психомоторного развития. При дальнейшем прогрессировании заболевания возникают эпилептические припадки, гиперкинезы, дыхательные расстройства. Диагностика синдрома Ли осуществляется на основании данных настоящего статуса больного, магнитно-резонансной томографии, молекулярно-генетических анализов. Специфического лечения данной патологии не существует, симптоматическая терапия лишь незначительно замедляет прогрессирование заболевания.
Общие сведения
Синдром Ли (подострая некротизирующая энцефаломиелопатия) – наследственное нейродегенеративное заболевание центральной нервной системы, которое характеризуется ранним началом и неуклонным прогрессированием неврологических нарушений. Впервые данное состояние было описано в 1951 году английским психиатром Денисом Ли, который определил его как наследственный вариант энцефаломиелопатии. Дальнейшие исследования показали, что синдром Ли является крайне гетерогенным состоянием с точки зрения этиологии – его причиной становятся дефекты множества генов, расположенных на аутосомах, Х-хромосоме и митохондриальной ДНК. По этой причине механизм наследования заболевания может быть (в зависимости от характера мутации) аутосомно-рецессивным, сцепленным с полом или митохондриальным. Из-за разнообразия генетических дефектов, являющихся причиной синдрома Ли, различается и половое распределение этого состояния, однако, по мнению многих врачей-генетиков, в целом можно считать, что оно в равной степени поражает как мальчиков, так и девочек. Встречаемость составляет ориентировочно 1 случай на 34-36 тысяч новорожденных.
Причины и классификация синдрома Ли
Причинами развития синдрома Ли могут выступать мутации широкого спектра генов, расположенных на разных хромосомах. Однако патогенез этого состояния примерно сходен у различных форм патологии и чаще всего связан с нарушением процессов клеточного дыхания и функционирования дыхательной цепи митохондрий. В отношении некоторых форм синдрома Ли также замечено нарушение функционирования пируватдегидрогеназного комплекса. Нарушение структуры белков дыхательной цепи митохондрий приводит к недостаточному синтезу АТФ, являющемуся основным источником энергии во всех клетках организма. Нейроны и клетки нейроглии особенно чувствительны к недостатку энергии, что становится причиной развития разнообразных нарушений еще с детского возраста. Классификация всех генетических дефектов при синдроме Ли основана на том, какой компонент дыхательной цепи (представляющей собой белковый комплекс) митохондрий нарушен в результате мутации.
В качестве отдельного варианта синдрома Ли часто указывают форму заболевания, обусловленную мутациями гена PDHA1, который расположен на Х-хромосоме. В результате наследование данного типа патологии является сцепленным с полом – болеют почти исключительно мальчики, тогда как женщины выступают носительницами патологических генов. Митохондриальный тип наследования синдрома Ли также имеет множество особенностей. Передача патологических генов происходит от матери к потомству и продолжается только по женской линии. Поскольку каждая митохондрия имеет собственную молекулу ДНК, в клетке одновременно присутствуют как «здоровые», так и «больные» органеллы, а при делении клеток (в том числе и при мейозе в процессе образования яйцеклеток) распределение больных генов оказывается неодинаковым. Женщины с относительно небольшим процентом «больных» митохондрий в клетках могут быть фенотипически здоровыми, но передавать их своему потомству. Невозможно точно предсказать, какое количество патологических митохондрий получит ребенок таких носителей, поэтому вероятность развития синдрома Ли у детей этих женщин неопределенная.
Симптомы
Проявления синдрома Ли обычно возникают на протяжении первого года жизни ребенка, иногда они могут регистрироваться в возрасте 2-5 лет, в редких случаях развитие заболевания начинается в подростковый период. Обычно первым проявлением патологии становится сонливость или, наоборот, повышенная возбудимость ребенка, у грудных детей наблюдается нарушение питания, недобор массы тела. В дальнейшем синдром Ли приводит к задержке психофизического развития, а у детей старшего возраста – к постепенной утрате уже обретенных навыков. Среди других неврологических симптомов заболевания наиболее часто отмечаются парезы, тремор конечностей, нарушение координации движения, поражение периферических нервов, снижение сухожильных рефлексов. В дальнейшем могут регистрироваться клонические судороги и эпилептические припадки.
Из-за недостатка энергии, обусловленного синдромом Ли, страдает не только нервная система, но и другие органы с высоким потреблением АТФ. В большинстве случаев у больных детей отмечается мышечная гипотония и слабость. Затрагивает заболевание и печень – орган с очень высоким потреблением энергии. У пациентов с синдромом Ли нередко выявляется увеличение печени, желтуха, иногда гепатолиенальный синдром. По мере прогрессирования патологии возникают нарушения дыхания – оно становится затрудненным, иногда приобретает характер дыхания Чейна-Стокса. У ряда больных со временем развивается миокардиодистрофия.
Синдром Ли имеет прогрессирующие течение. На терминальных этапах наблюдается поражение органов зрения, которое проявляется нистагмом, нарушением цветовосприятия, косоглазием. В конечном итоге может возникнуть атрофия зрительного нерва и полная слепота. Мышечная гипотония и гипорефлексия сменяются спастическим напряжением мышц и повышением сухожильных рефлексов. Через 2-7 лет после появления первых симптомов синдрома Ли происходит резкое падение массы тела, вышеперечисленные проявления резко усиливаются, наступает летальный исход по причине дыхательной или сердечно-сосудистой недостаточности.
Диагностика и лечение синдрома Ли
Для определения наличия синдрома Ли применяют магнитно-резонансную томографию головного мозга, электронейромиографию, изучение наследственного анамнеза, молекулярно-генетические анализы. При осмотре выявляют характерные неврологические симптомы, тремор конечностей, отставание в психофизическом развитии, у младенцев – недобор массы тела. На магнитно-резонансной томографии мозга обнаруживают симметричные изменения в области продолговатого мозга, таламуса и моста, иногда аналогичные изменения могут регистрироваться и в спинном мозге. Наилучшие результаты диагностики синдрома Ли при помощи МРТ получаются при использовании T2W и FLAIR режимов.
В тех случаях, когда имеются признаки поражения периферических нервов и мышц, для диагностики синдрома Ли выполняют электронейромиографию. При этом заболевании главным и наиболее частым результатом ЭНМР становится замедление скорости прохождения нервного импульса, которое свидетельствует о демиелинизации нервов. Изучение наследственного анамнеза информативно в случае аутосомно-рецессивных форм заболевания, при мутации генов митохондриальной ДНК четко определить семейный характер патологии затруднительно. Молекулярно-генетическая диагностика массово используется только в отношении некоторых форм синдрома Ли (обусловленных мутациями генов BCS1L, SURF1 и некоторых других).
Специфического лечения данной патологии не существует, применяется симптоматическая терапия: противосудорожные и ноотропные средства, препараты для улучшения мозгового кровообращения. Важную роль в лечении синдрома Ли играет назначение витаминов, служащих кофакторами ферментов дыхательной цепи митохондрий – В1, В6, Q10. Их регулярный прием позволяет несколько замедлить прогрессирование заболевания и уменьшить выраженность симптомов. Однако, несмотря на все предпринятые терапевтические меры, 80% больных умирает через 2-7 лет после регистрации первых проявлений патологии.
Прогноз и профилактика
Прогноз синдрома Ли крайне неблагоприятный, так как большинство больных умирает через несколько лет после возникновения заболевания. Симптоматическое лечение может несколько замедлить прогрессирование патологии и ослабить выраженность проявлений, однако полноценного улучшения оно не обеспечивает. В большинстве случаев еще за год-два до летального исхода наступает полная инвалидизация больного, обусловленная неврологическими, дыхательными и метаболическими нарушениями. Причиной смерти при синдроме Ли чаще всего становится сердечно-сосудистая или дыхательная недостаточность. Профилактика этого заболевания осуществляется в рамках медико-генетического консультирования родителей перед зачатием ребенка.



