Амиотрофия Верднига-Гоффмана — это наиболее злокачественная спинальная мышечная атрофия, развивающаяся с рождения или в первые 1-1,5 года жизни ребенка. Характеризуется нарастающими диффузными мышечными атрофиями, сопровождающимися вялыми парезами, прогрессирующими до полной плегии. Как правило, амиотрофия Верднига-Гоффмана сочетается с костными деформациями и врожденными аномалиями развития. Диагностическую основу составляет анамнез, неврологический осмотр, электрофизиологические и томографические исследования, анализ ДНК и изучение морфологического строения мышечной ткани. Лечение слабо эффективно, направлено на оптимизацию трофики нервной и мышечной тканей.
МКБ-10
Общие сведения
Амиотрофия Верднига-Гоффмана является самым тяжелым вариантом из всех спинальных мышечных атрофий (СМА). Ее распространенность находится на уровне 1 случай на 6-10 тыс. новорожденных. Заболевание имеет несколько форм: врожденную, промежуточную (раннюю детскую) и позднюю. Целый ряд специалистов выделяет последнюю форму как самостоятельную нозологию — амиотрофию Кугельберга-Веландера. Отсутствие этиотропного и патогенетического лечения, ранний летальный исход ставят курирование пациентов с болезнью Верднига-Гоффмана в ряд наиболее сложных задач, стоящих перед современной неврологией и педиатрией.
Причины
Амиотрофия Верднига-Гоффмана — наследственная патология, кодируемая поломкой в генетическом аппарате на уровне локуса 5q13 5-й хромосомы. Ген, в котором происходят мутации, получил название survival motor neuron gene (SMN) — ген, ответственный за выживание мотонейронов. У 95% пациентов с болезнью Верднига-Гоффмана отмечается делеция теломерной копии этого гена. Тяжесть СМА прямо коррелирует с протяженностью участка делеции и сопутствующим наличием изменений (рекомбинации) в генах H4F5, NAIP, GTF2H2.
Носителем измененного гена, обуславливающего возникновение заболевания, является каждый 50-й человек. Но благодаря аутосомно-рецессивному типу наследования, патология у ребенка проявляется только тогда, когда соответствующая генетическая аберрация имеется и у матери, и у отца. Вероятность рождения ребенка с патологией в такой ситуации составляет 25%.
Патогенез
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах. Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты. Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
Симптомы амиотрофии
Врожденная форма
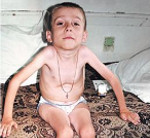
СМА I клинически манифестирует до 6-месячного возраста. Внутриутробно может проявляться вялым шевелением плода. Зачастую мышечная гипотония отмечается с первых дней жизни и сопровождается угасанием глубоких рефлексов. Дети слабо кричат, плохо сосут, не могут держать голову. В отдельных случаях (при более позднем дебюте симптомов) ребенок учится держать голову и даже сидеть, но на фоне развития заболевания эти навыки быстро исчезают. Характерны ранние бульбарные нарушения, понижение глоточного рефлекса, фасцикулярные подергивания языка.
Данная амиотрофия Верднига-Гоффмана сочетается с олигофренией и нарушениями формирования костно-суставного аппарата: деформациями грудной клетки (воронкообразной и килевидной грудной клеткой), искривлением позвоночника (сколиозом), контрактурами суставов. У многих пациентов выявляются другие врожденные аномалии: гемангиомы, гидроцефалия, косолапость, дисплазия тазобедренных суставов, крипторхизм и пр.
Течение СМА I наиболее злокачественное с быстро нарастающей обездвиженностью и парезом дыхательной мускулатуры. Последний обуславливает развитие и прогрессирование дыхательной недостаточности, выступающей основной причиной летального исхода. В связи с нарушением глотания возможен заброс пищи в дыхательные пути с развитием аспирационной пневмонии, которая может явиться смертельно опасным осложнением спинальной амиотрофии.
Ранняя детская форма
СМА II дебютирует после 6-месячного возраста. К этому периоду дети имеют удовлетворительное физическое и нервно-психическое развитие, в соответствии с возрастными нормами приобретают навыки держать голову, переворачиваться, садиться, стоять. Но в подавляющем большинстве клинических случаев дети так и не успевают научиться ходить. Обычно эта амиотрофия Верднига-Гоффмана манифестирует после перенесенной ребенком пищевой токсикоинфекции или другого острого инфекционного заболевания.
В начальном периоде периферические парезы возникают в нижних конечностях. Затем они достаточно быстро распространяются на верхние конечности и мускулатуру туловища. Развивается диффузная мышечная гипотония, происходит угасание глубоких рефлексов. Наблюдаются контрактуры сухожилий, тремор пальцев, непроизвольные мышечные сокращения (фасцикуляции) языка. На поздних стадиях присоединяются бульбарные симптомы, прогрессирующая дыхательная недостаточность. Течение более медленное, чем у врожденной формы болезни Верднига-Гоффмана. Пациенты могут доживать до 15-летнего возраста.
Амиотрофия Кугельберга-Веландера
Диагностика
Больные спинальной мышечной атрофией I типа находятся под наблюдением детских неврологов и неонатологов. Большое значение имеет возраст манифестации заболевания – для амиотрофии Верднига-Гоффмана характерно развитие с момента рождения до 6 месяцев. В анамнезе часто имеются сведения о позднем и вялом шевелении плода во время беременности.
При осмотре ребенка обращается внимание на выраженную мышечную гипотонию, неспособность самостоятельно сидеть или удерживать голову, типичную «позу лягушки» – плечи приподняты, руки вдоль туловища, ноги выпрямлены и повернуты кнаружи. Отмечаются мышечные подергивания, ослабление или отсутствие сухожильных рефлексов, грубые костные деформации (колоколообразная грудная клетка, Х-образные нижние конечности). Для подтверждения диагноза назначаются следующие дополнительные исследования:
Амиотрофию Верднига-Гоффмана следует дифференцировать с другими генетически обусловленными нервно-мышечными заболеваниями, имеющими такое же быстропрогрессирующее течение. К ним относятся врожденные структурные миопатии, ювенильный боковой амиотрофический склероз, синдром Фукуямы.
Лечение амиотрофии Верднига-Гоффмана
Немедикаментозная терапия
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В тяжелых ситуациях (например, при выраженной гипоксемии вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. На сегодняшний день не существует этиотропной терапии амиотрофии Верднига-Гоффмана. Все мероприятия носят симптоматический и паллиативный характер. Применяются следующие методы лечения:
Медикаментозная терапия
Для достижения максимального эффекта лечение должно быть комплексным, проводиться непрерывно и подбираться индивидуально для конкретного пациента. Лекарственные препараты, применяющие для лечения амиотрофии Верднига-Гоффмана следующие:
Хирургическое лечение
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. У лежачих больных, страдающих постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Новейшие разработки
Ведутся постоянные исследовательские работы по поиску эффективного лекарства для терапии спинальной мышечной атрофии. Наиболее перспективным направлением считается генная терапия. В клинической практике уже используются антисмысловые олигонуклеотиды, исправляющие дефекты матричной РНК в гене SMN2 (Спинраза).
В конце 2019 года был зарегистрирован и одобрен к клиническому применению препарат Золгенсма, который содержит функционально полноценный ген SMN1. Доставка этого гена в нервные клетки производится с помощью аденоассоциированного вируса, проникающего через гематоэнцефалический барьер. Использование Золгенсмы приводит к значительному увеличению продукции белка SMN1 и улучшению состояния пациентов.
Прогноз
Врожденная амиотрофия Верднига-Гоффмана имеет крайне неблагоприятный прогноз. При ее манифестации в первые дни жизни ребенка, его гибель, как правило, происходит до 6-месячного возраста. При начале клиники после 3-х месяцев жизни, летальный исход наступает в среднем к возрасту 2 года, иногда — к 7-8 годам. Ранняя детская форма характеризуется более замедленным прогрессированием, дети погибают в возрасте 14-15 лет.
Профилактика
Первичная профилактика амиотрофии Верднига-Гоффмана заключается в пренатальной диагностике. При обнаружении в биоптатах ворсин хориона или амниотической жидкости мутации SMN1 показано прерывание беременности. Вторичная профилактика сводится к предупреждению осложнений – аспирационной пневмонии, контрактур суставов, инфекций нижних дыхательных путей.
Почему у людей бывает СМА?



Спинальная мышечная атрофия – это заболевание, при котором повреждается ген SMN1. В результате мутации у человека не вырабатывается белок, который нужен для выживания мотонейронов. Мотонейроны — важная часть спинного мозга человека, от них по длинным нервам идут сигналы к скелетным мышцам организма. Если мотонейрон получает недостаточно белка для своего питания, то он быстро устает. Ему приходится работать и за себя, и «за того парня». Многие мотонейроны просто гибнут от «усталости», — нагрузка на оставшиеся возрастает, и в результате они работают с чудовищной перегрузкой. От усталости они не могут посылать сигналы к скелетным мышцам, благодаря которым человек ходит, сидит, лежит, глотает или дышит. Мышцы остаются без работы, потому что их никто ничего не просит сделать, и постепенно отмирают.
Дефицит важного белка, который не вырабатывает ген SMN1, приводит к атрофии (потере жизнеспособности и уменьшению размера) мышц, и в результате человек постепенно теряет способность ходить, удерживать и управлять собственным телом, самостоятельно сидеть, есть, глотать и дышать. Упрощенный механизм СМА можно увидеть на схеме.

К счастью, у человека есть еще ген SMN2, который дублирует SMN1. Он производит нефункциональный белок и небольшое количество нормального белка, который позволяет мотонейронам человека выживать на очень голодном пайке. В результате человек со СМА сохраняет какую то часть своих двигательных, глотательных и дыхательных функций.
Спинальные амиотрофии ( Спинальные мышечные атрофии )

Спинальные амиотрофии — это генетические заболевания, проявляющиеся мышечной атрофией и обусловленные дегенеративными изменениями спинальных мотонейронов и моторных ядер ствола головного мозга. Общим симптомокомплексом выступают симметричные вялые параличи с атрофиями мышц и фасцикуляциями на фоне интактной чувствительной сферы. Диагностируются спинальные амиотрофии по данным семейного анамнеза, неврологического статуса, ЭФИ нервно-мышечного аппарата, МРТ позвоночника, ДНК-анализа и морфологического исследования мышечного биоптата. Лечение малоэффективно. Прогноз зависит от формы спинальной мышечной атрофии и возраста ее дебюта.

Общие сведения
Спинальные амиотрофии (спинальные мышечные атрофии, СМА) — наследственно обусловленные заболевания, в основе которых лежит дегенерация мотонейронов спинного мозга и ствола головного мозга. Описаны в конце XIX века. Их частота составляет 1 случай на 6-10 тыс. новорожденных. Около 85% спинальных мышечных атрофий составляют проксимальные формы с более выраженной слабостью и атрофиями проксимальных мышечных групп конечностей. На долю дистальных форм приходится лишь 10% СМА. На сегодняшний день спинальные амиотрофии представляют практический интерес для целого ряда дисциплин: детской и взрослой неврологии, педиатрии, генетики.
Причины
Благодаря современной генетике установлено, что возникающие дегенеративные процессы двигательных нейронов обусловлены мутациями в генах SMN, NAIP, H4F5, ВTF2p44, расположенных на 5-ой хромосоме в локусе 5q13. Несмотря на то, что спинальные амиотрофии детерминируются аберрациями одного хромосомного локуса, они представляют собой группу разнородных нозологий, одни из которых проявляются в младенческом возрасте, а другие манифестируют у взрослых. В большинстве случаев амиотрофии наследуются аутосомно-рецессивно.
Патогенез
Генетические мутации приводят к развитию дегенеративных изменений в передних рогах спинного мозга. Нарушается иннервация и нейротрофика поперечно-полосатой мускулатуры. В результате постепенно возникает атрофия мышечной ткани. Преимущественное поражение отдельных групп мышц (проксимальных или дистальных частей верхних или нижних конечностей) у различных форм спинальных амиотрофий отличается. Характерно отсутствие нарушений чувствительности.
Классификация
Общепринятым считается разделение спинальных мышечных атрофий на детские и взрослые. Детские СМА классифицируются на ранние (дебютирующие в первые месяцы жизни), более поздние и ювенильные. Детские спинальные амиотрофии представлены:
Взрослые формы СМА манифестируют в возрасте от 16 до 60 лет и отличаются более доброкачественным клиническим течением. К СМА взрослого возраста относятся:
Выделяют также изолированные и сочетанные спинальные амиотрофии. Изолированные СМА характеризуются преобладанием поражения спинальных мотонейронов, которое во многих случаях является единственным проявлением заболевания. Сочетанные спинальные амиотрофии представляют собой редкие клинические формы, при которых симптомокомплекс амиотрофии комбинируется с другой неврологической или соматической патологией. Описаны сочетания СМА с врожденными пороками сердца, глухотой, олигофренией, понтоцеребеллярной гипоплазией, врожденными переломами.
Симптомы спинальных амиотрофий
Общим для спинальных мышечных атрофий является симптомокомплекс симметричного вялого периферического паралича: слабость, атрофия и гипотония мышечных групп одноименных конечностей (чаще вначале обеих ног, а затем и рук) и туловища. Пирамидные нарушения не типичны, но могут развиваться на поздних стадиях. Расстройства чувствительности отсутствуют, функция тазовых органов сохранена. Обращает внимание более выраженное поражение проксимальных (при проксимальных СМА) или дистальных (при дистальных СМА) мышечных групп. Типично наличие фасцикулярных подергиваний и фибрилляций.
Болезнь Верднига-Гоффмана
Встречается в 3-х клинических вариантах. Врожденный вариант дебютирует в первые 6 мес. жизни и является наиболее злокачественным. Его симптомы могут проявляться еще во внутриутробном периоде слабым шевелением плода. Дети с рождения имеют мышечную гипотонию, не способны переворачиваться и держать голову, при более позднем дебюте — не могут сидеть. Патогномонична поза лягушки — ребенок лежит с разведенными в стороны и согнутыми в коленях и локтях конечностями.
Ранняя спинальная амиотрофия манифестирует до 1,5 лет зачастую после инфекционного заболевания. Ребенок утрачивает двигательные способности, не может стоять и даже сидеть. Периферические парезы сочетаются с контрактурами. После вовлечения дыхательных мышц развивается дыхательная недостаточность и застойная пневмония. Летальный исход обычно происходит в возрасте до 5-ти лет. Поздний вариант дебютирует после 1,5 лет, отличается сохранением двигательной способности до 10-летнего возраста. Летальный исход наступает к 15-18 годам.
Ювенильная спинальная амиотрофия Кугельберга-Веландера
Характеризуется дебютом в период от 2 до 15 лет. Начинается с поражения проксимальных мышц ног и тазового пояса, затем захватывает плечевой пояс. Около четверти пациентов имеют псевдогипертрофии, что делает клинику сходной с проявлениями мышечной дистрофии Беккера. В плане дифдиагностики большое значение имеет наличие мышечных фасцикуляций и данные ЭМГ. Течение амиотрофии Кугельберга-Веландера доброкачественное без костных деформаций, в течение ряда лет пациенты остаются способными к самообслуживанию.
Бульбоспинальная амиотрофия Кеннеди
Наследуется рецессивно сцеплено с Х-хромосомой, манифестирует только у мужчин после 30-летнего возраста. Типично медленное, относительно доброкачественное течение. Дебютирует с амиотрофии проксимальных мышц ног. Бульбарные расстройства появляются через 10-20 лет и благодаря медленному прогрессированию не вызывают нарушения витальных функций. Может наблюдаться тремор головы и рук. Патогномоничным симптомом выступают фасцикулярные подергивания в периоральных мышцах. Зачастую отмечается эндокринная патология: атрофия яичек, снижение либидо, гинекомастия, сахарный диабет.
Дистальная СМА Дюшенна-Арана
Может иметь как рецессивный, так и доминантный тип наследования. Дебют приходится чаще на 20-летний возраст, но может произойти в любой период до 50 лет. Амиотрофии начинаются в кистях рук и приводят к формированию «когтистой кисти», затем охватывают предплечье и плечо, в связи с чем рука приобретает вид «руки скелета». Парезы мышц голеней, бедер и туловища присоединяются гораздо позже. Описаны случаи манифестации заболевания монопарезом (поражением одной руки). Прогноз благоприятный, за исключением случаев сочетания данного вида СМА с торсионной дистонией и паркинсонизмом.
Скапуло-перонеальная СМА Вюльпиана
Манифестирует в период от 20 до 40 лет амиотрофиями плечевого пояса. Типичны «крыловидные лопатки». Затем присоединяется поражение перонеальной группы мышц (разгибатели стопы и голени). В ряде случаев вначале поражаются перонеальные мышцы, а затем плечевой пояс. Спинальная амиотрофия Вюльпиана отличается медленным течением с сохранностью способности передвигаться спустя 30-40 лет от ее дебюта.
Осложнения
К наиболее частым неблагоприятным последствиям можно отнести постоянные падения и ассоциированные с ними патологические переломы, что связано с постепенно нарастающей мышечной атрофией мышц нижних конечностей. При бульбоспинальной амиотрофии Кеннеди нередко наблюдаются осложнения со стороны эндокринной и репродуктивной систем – эректильная дисфункция, первичное бесплодие, сахарный диабет.
Более тяжелые состояния встречаются реже, в основном при болезни Верднига-Гоффмана или на поздних стадиях других амиотрофий. Вследствие выраженной атрофии мышц глотки и дыхательной мускулатуры пища попадает в дыхательные пути (аспирация), развивается дыхательная недостаточность. Возможны грубые костно-суставные деформации, утрата способности к ходьбе и самообслуживанию. В единичных случаях обнаруживается рак грудной железы.
Диагностика
Больных со спинальными амиотрофиями курируют врачи-неврологи, а при манифестации в детском возрасте – педиатры или неонатологи. При необходимости может потребоваться привлечение для консультации других специалистов – эндокринологов и генетиков. Для диагностики некоторых разновидностей амиотрофий большое значение имеют анамнестические данные, а именно возраст появления симптоматики (например, болезнь Верднига-Гоффмана всегда дебютирует у детей до 6 месяцев, а амиотрофия Кугельберга-Веландера – после 2 лет).
Во время осмотра пациента обращается внимание на снижение общего мышечного тонуса, ослабление или утрату сухожильных рефлексов, скелетно-мышечные деформации. У части больных отмечается псевдогипертрофия икроножных мышц. Чтобы подтвердить или исключить диагноз назначается следующее дополнительное обследование:
Дифференциальная диагностика
Дифференциальный диагноз спинальных амиотрофий необходимо проводить с другими наследственными нейродегенеративными заболеваниями, поражающими мышечную ткань. К таким патологиям можно отнести:
Лечение спинальных амиотрофий
Немедикаментозная терапия
Все без исключения больные должны быть госпитализированы в стационар. В тяжелых ситуациях (например, при дыхательной недостаточности вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. Все мероприятия направлены на облегчение состояния пациента. Несмотря на это, комплексный подход и строгое соблюдение рекомендаций врачей позволяют улучшить качество жизни больного. Виды консервативной терапии, применяющиеся для терапии спинальных амиотрофий:
Для достижения максимального эффекта лечение должно проводиться непрерывно и подбираться индивидуально для конкретного пациента.
Медикаментозная терапия
Этиотропная терапия с доказанной эффективностью на сегодняшний день отсутствует. Лекарственные препараты, применяющие для лечения спинальных амиотрофий, следующие:
Хирургическое лечение
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. Лежачим больным, страдающим постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Экспериментальное лечение
Постоянно ведутся многочисленные исследовательские работы по разработке лекарственных средств, способных остановить или хотя бы замедлить процесс нейродегенерации. Значительные успехи достигнуты в области генной терапии. Уже используются в клинической практике препараты, корректирующие дефекты матричной РНК гена SMN – Спинраза и Рисдиплам. В конце 2019 года был зарегистрирован и одобрен к клиническому применению препарат Золгенсма, который содержит функционально полноценный ген SMN1.
Прогноз
Прогноз всецело зависит от клинического варианта СМА и возраста ее манифестации. Наиболее неблагоприятный прогноз имеют детские спинальные амиотрофии, при начале в младенческом возрасте они зачастую приводят к летальному исходу в течение первых 2-х лет жизни ребенка. Спинальные амиотрофии взрослого возраста отличаются способностью больных самостоятельно обслуживать себя в течение многих лет, а при медленном прогрессировании имеют благоприятный прогноз не только для жизни, но и для трудоспособности пациентов (при создании для них оптимальных условий труда).
Профилактика
Специфических методов первичной профилактики не существует. Единственный способ предотвратить возникновение болезни – пренатальная диагностика (обнаружение мутаций в ворсинах хориона или амниотической жидкости) с прерыванием беременности. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Как диагностируют и лечат спинальную мышечную атрофию

О спинальной мышечной атрофии (СМА) — тяжелом редком генетическом заболевании — стали активно говорить в обществе и СМИ лишь несколько лет назад, хотя само заболевание, конечно, известно науке давно. Впервые оно было описано в 1891 году австрийским неврологом Г. Верднигом и затем в 1892 году немецким неврологом Й. Гоффманом.
Сегодня оживление вокруг проблематики СМА вызвано тем, что лишь относительно недавно благодаря активному развитию фармацевтической промышленности и генной терапии появились препараты, способные существенно замедлить прогрессирование заболевания. До этого момента болезнь считалась неизлечимой, а постановка диагноза «СМА» была равноценна приговору.
В последние годы о СМА рассказывают пациентские и благотворительные организации, медицинские работники, СМИ, а отдельные люди и фонды собирают средства на покупку дорогостоящих лекарств для таких больных. В результате самое общее представление о СМА имеют и многие обычные люди, далекие от медицины. Например, знают, что оно связано с тяжелой инвалидностью, сильным ограничением подвижности и что цены на спасительные лекарства от СМА исчисляются десятками миллионов рублей.
Что сегодня известно о СМА науке? Почему лечение этой болезни так дорого стоит? Как сегодня живут пациенты с этим диагнозом? Давайте разбираться.

Автор статьи — Кристина Невмержицкая, врач-невролог, заведующая отделением неврологии Областной детской клинической больницы Екатеринбурга, медицинский эксперт благотворительного фонда «Семьи СМА»
Что такое СМА
СМА — это аутосомно-рецессивное нервно-мышечное заболевание, характеризующееся прогрессирующими симптомами вялого паралича и мышечной атрофии. СМА одно из самых распространенных редких (орфанных) заболеваний. Распространенность СМА составляет 1 на 6–10 тыс. новорожденных. Согласно официальным данным, в России сегодня 890 детей со СМА. Каждый год рождается около 200 детей с этим диагнозом [1].
При СМА из-за генетической поломки возникает недостаток особого белка SMN (survival motor neuron). Это провоцирует постепенную гибель α-мотонейронов спинного мозга, которые иннервируют скелетные мышцы и отвечают за их сокращение.
В синтезе белка SMN участвуют два гена — SMN1 и SMN2. Ген SMN1 основной, он кодирует 85% белка. Второй ген SMN2 — оставшиеся 15% белка. Таким образом, если из-за мутации ген SMN1 не работает, единственным источником информации о белке SMN становится ген SMN2. Однако у больных СМА разное количество копий гена SMN2. Чем больше таких копий, кодирующих белок SMN, тем, как правило, легче течение болезни и благоприятнее прогноз. Количество копий гена SMN2 устанавливается путем генетического анализа.
Глобально у всех больных СМА наблюдается прогрессирующая мышечная слабость, ухудшение или постепенная потеря амплитуды движений, нарушения дыхания и глотания. При этом у больных сохраняется интеллект, способность к обучению и выполнению профессиональных навыков.
Как наследуется СМА
СМА — генетическое заболевание с аутосомно-рецессивным типом наследования. Это значит, что родители ребенка со СМА могут быть носителями дефектного гена SMN1, при этом заболевание не проявляется, а сами они могут не подозревать о риске передать его потомству. В случае если оба родителя передадут поврежденный ген ребенку, СМА разовьется у него с вероятностью 25%.
Если же ребенок получит поврежденный ген только от одного родителя, то не будет иметь симптомов заболевания, а так же, как и родители, будет носителем этого дефектного гена. Вероятность этого составляет 50%.

Типы СМА
В настоящее время принято выделять несколько типов спинальной мышечной атрофии на основании возраста появления первых симптомов и максимальных двигательных умений больного. Каждый тип при естественном течении заболевания связан с разной прогнозируемой продолжительностью жизни. По результату генетического анализа тип СМА установить нельзя.
СМА не зря называют коварным заболеванием. Нередко его признаки появляются внезапно, на фоне нормального общего развития, после чего все приобретенные навыки — удержания головы, сидения, ползания, стояния — постепенно теряются.
Классических типов СМА пять. Отдельно выделяют СМА 0 и IV типов [2].
СМА 0
СМА 0 типа нередко объединяют с I типом СМА. Он также, как и I тип, имеет самое тяжелое течение. СМА 0 типа проявляется еще внутриутробно — ребенок начинает менее активно шевелиться. С первых дней жизни у новорожденного наблюдаются резко сниженная спонтанная двигательная активность и мышечная гиподинамия, отсутствие сухожильных рефлексов, возникают проблемы с дыханием и глотанием.
Продолжительность жизни детей с этим типом СМА составляет несколько недель.
СМА I
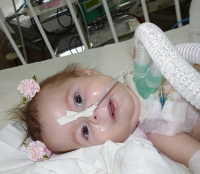
Самый тяжелый тип СМА. Первые признаки заболевания возникают в возрасте до шести месяцев. Как правило, родители первыми замечают тревожные симптомы: ребенок не держит голову, не переворачивается и не может сидеть без посторонней поддержки. Дети со СМА I типа очень слабые, не могут в полную силу кричать и кашлять, испытывают трудности с принятием пищи, глотанием, дыханием и отхождением мокроты, в результате чего у них высок риск осложненного течения респираторных заболеваний.
Ожидаемая продолжительность жизни таких больных составляет меньше двух лет. Более ⅔ детей со СМА не доживают до двух лет и погибают в результате развития дыхательной недостаточности из-за слабости дыхательной мускулатуры.
СМА II
Данный тип СМА протекает медленнее и впервые проявляется в возрасте от шести до 18 месяцев. Главный признак — ребенок с трудом встает на ноги, в большинстве случаев у него вовсе не получается удерживать вертикальное положение тела. Ходить такие дети не могут. Также наблюдается задержка моторного развития, дрожание рук, сколиоз вследствие слабости мышц и контрактуры суставов. Больным может потребоваться инвалидная коляска. Вкупе с другими приспособлениями — поддержками, упорами — это позволяет зафиксировать оптимальное положение тела пациента.
Ожидаемая продолжительность жизни больных с этим типом СМА — 20–40 лет.

СМА III
Для третьего типа СМА характерно стабильное развитие больных до определенного возраста. Они могут сидеть, стоять и ходить. Затем болезнь проявляет себя — из-за прогрессирующей слабости мышц нижних конечностей походка постепенно становится неустойчивой. В ряде случае пациенты теряют способность ходить.
Продолжительность жизни больных со СМА III типа, как правило, такая же, как у обычных людей.
СМА IV
Этот тип СМА считается самым легким. Он проявляется уже во взрослом возрасте — после 25 лет. У таких больных наблюдаются умеренные двигательные нарушения, мышечная слабость, тремор. В ряде случаев из-за прогрессирования заболевания теряется способность ходить.
Диагностика СМА
Крайне важно установить диагноз как можно раньше, чтобы начать лечение и реабилитацию [3], [4].
Для подтверждения диагноза обычно проводят молекулярно-генетический анализ. Он показывает, имеется ли у больного дефект в гене SMN1, а также число копий гена SMN2. Анализ может быть назначен любым врачом, заподозрившим СМА. Педиатр и невролог может заметить настораживающее отставание развития ребенка от возрастных норм, поэтому родителям важно вовремя проводить все плановые осмотры ребенка и самим наблюдать за его физическим развитием.
При необходимости могут быть назначены и другие исследования, в том числе дополнительный генетический анализ, электромиография, некоторые лабораторные исследования, которые в большей степени служат для исключения похожих заболеваний. После уточнения диагноза требуется осмотр несколькими узкими специалистами для оценки факторов риска развития дыхательной недостаточности, ортопедических проблем, трудностей кормления и откашливания.
Очень важно, чтобы с каждым больным ребенком работала мультидисциплинарная команда специалистов, знающих все аспекты и нюансы лечения и реабилитации больных СМА.
Лечение СМА
Сегодня для лечения СМА в мире применяются три препарата.
Крупнейшие мировые фармацевтические компании параллельно исследуют и разрабатывают лекарства. В том числе оценивается эффективность увеличения доз препаратов, тестируются различные способы введения и комбинации лекарств.
Два из трех разработанных препаратов подходят для лечения всех типов СМА и на всех этапах. Однако чем раньше начинается лечение, тем лучшего эффекта можно достичь. Данная группа лекарств влияет на работу гена SMN2, который, в свою очередь, кодирует белок SMN, отвечающий за выживание нейронов спинного мозга. Препараты позволяют стабилизировать течение заболевания, улучшить двигательные функции больного и приобрести новые моторные навыки.
Третий революционный препарат для лечения СМА у детей — продукт генной терапии. При помощи вирус-вектора препарат доставляет синтетическую функциональную копию гена SMN1 в нейроны спинного мозга. В результате лечения в моторных нейронах, лишенных гена SMN1, восстанавливается продуцирование белка SMN. Дозировка этого лекарства устанавливается индивидуально для каждого пациента в зависимости от веса. Безопасность и эффективность препарата установлены у детей в возрасте до двух лет или с весом до 21 кг. Лекарство применяется однократно — вводится пациенту путем внутривенной инфузии в течение одного часа. Из-за дороговизны разработка стоимость такого лечения достигает 150 млн руб.
На сегодняшний день любая медикаментозная терапия пациентам со СМА должна назначаться специалистами, имеющими достаточный опыт в ее применении.
Жизнь со СМА: как помочь больному
До появления препаратов для лечения СМА больным оказывалась только паллиативная помощь, призванная максимально облегчить симптомы [5].
Теперь речь идет о реабилитации, однако отработанных эффективных методик пока мало. На данный момент с уверенностью можно говорить только об эффективности физической терапии и гидрокинезотерапии.
Огромную роль в реабилитации больного со СМА играют его близкие. Вместе в врачами разных профилей они образуют единую команду, совместными усилиями которой ребенку могут быть обеспечены максимально эффективная системная реабилитация и уход.
Физическая терапия
Из-за общей мышечной слабости больные СМА не могут совершать движения в том же объеме, что здоровые люди. Из-за этого возникает риск развития контрактур суставов, которые со временем могут стать постоянными и ограничивать движения. Лечебная физкультура включает комплексы упражнений по восстановлению гибкости и общих функциональных возможностей. Упражнения подбираются индивидуально для каждого пациента в зависимости от особенностей его здоровья. Например, для полностью лежачих больных занятия ставят целью в том числе увеличение объема и амплитуды активных/пассивных движений в любом сегменте тела и конечностей. У сидячих пациентов — повышение мобильности.
В лечебной физкультуре также применяются различные вспомогательные устройства и приспособления. Например, вертикализатор позволяет улучшить прочность костей и может использоваться в домашних условиях.
Гидрокинезотерапия
Гидрокинезотерапия представляет собой комплекс водных лечебных упражнений, дополняемый подводным массажем и ортопедическими приспособлениями.
Упражнения проходят в специально оснащенном бассейне при оптимальной температуре воды. В целом вода — это дружественная среда для больных СМА, в том числе за счет снятия нагрузки на позвоночник, суставы и сердце. Занятия позволяют укрепить все группы мышц, способствуют восстановлению двигательных функций, повышают общий тонус пациентов и очень нравятся детям.
Где узнать о СМА больше
SMA Europe — европейская пациентская организация, созданная в 2006 году. Она объединяет пациентские и исследовательские организации всей Европы, посвященные СМА.
«Семьи СМА» — российская пациентская организация, часть SMA Europe, созданная в 2015 году. На сайте представлена актуальная информация о СМА для пациентов и их семей, в том числе последние новости о законодательных аспектах и фармацевтическом рынке. Работает горячая линия.
TogetherInSMA— международный сайт, созданный для информирования пациентов со СМА и их семей. Здесь на русском языке можно получить общую информацию о СМА, доступных методах лечения, найти практические руководства по уходу за больными и другие полезные сведения.
Вниманиесма.рф — информационный портал, созданный при поддержке второй по величине в мире фармацевтической компании. Ресурс полностью посвящен симптоматике СМА, в том числе первым тревожным симптомам заболевания.
Узнать о том, как поддержать людей с редкими (орфанными) заболеваниями можно на сайте «Редкое искусство помогать» и в аккаунте @rare_art.
Важно помнить, что ни информация данных ресурсов, ни эта статья не заменяют очную консультацию у медицинского специалиста по любым вопросам, связанным с диагностикой и лечением СМА.



