Орфанные препараты — новая категория на фармрынке
Роза Ягудина о росте количества орфанных препаратов и их роли в лечении пациентов
Нередкие редкие (орфанные) заболевания
Редкие болезни известных людей
Примеры списка орфанных заболеваний
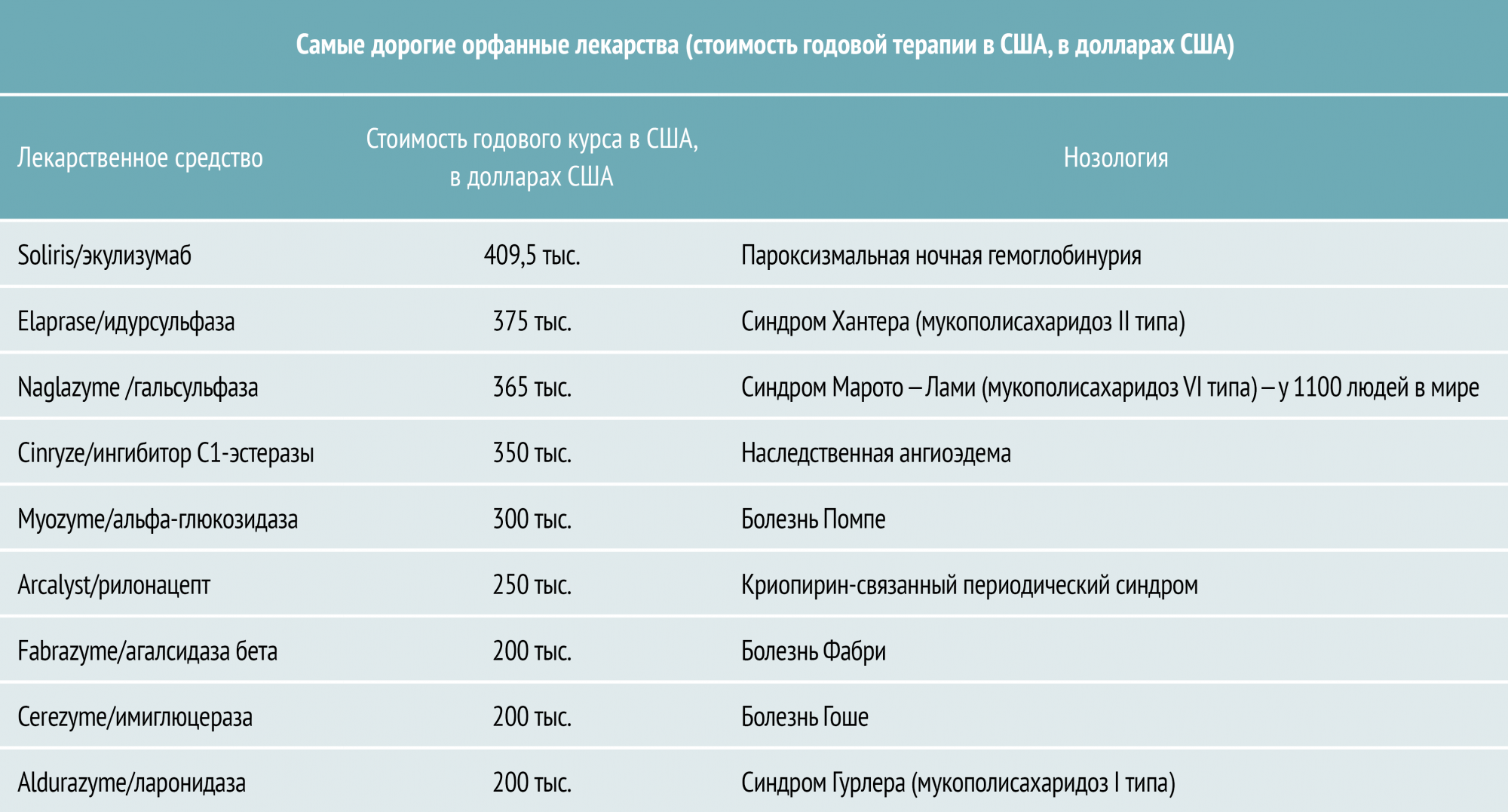
Почему так дорого?
Материалы об орфанных препаратах и заболеваниях:
Системные васкулиты как пример редких, орфанных заболеваний
Первичные системные васкулиты — редкие (орфанные) заболевания, и врачи, работающие в обычных лечебных учреждениях, а не в специализированных центрах, просто не могут накопить необходимого опыта в распознавании и лечении этих болезней.

Новые открытия и актуальные проблемы в мире лекарств
Сегодня орфанным препаратам стали уделять повышенное внимание. Появились препараты для лечения таких редких болезней, как мукополисахаридоз II типа, болезнь Ниманна-Пика и так далее.

Оригинальные лекарственные препараты и дженерики
В «лекарственном портфеле» многих инновационных компаний присутствуют орфанные препараты, которые окупаются долго и не приносят большой прибыли. Их разработка и производство являются следствием осознания ответственности перед каждым пациентом в отдельности.
Нашли ошибку? Выделите текст и нажмите Ctrl+Enter.


Редакция не несет ответственности за информацию, размещенную в рекламных материалах. Мнение редакции может не совпадать с мнением наших авторов. Все материалы, опубликованные в журнале, охраняются законом «Об авторском праве». Любое воспроизведение статей, перепечатка либо ссылка на них допускаются исключительно с письменного согласия редакции.
Сирота казенная

РЕДКОСТНЫЙ ГЕН
На сегодняшний день, по данным The European Organisation for Rare Diseases, в мире насчитывается 6–8 тысяч орфанных заболеваний. Порядка 80% этих патологий считаются генетическими, первые симптомы могут проявляться уже при рождении или в раннем возрасте. В большинстве своем это тяжелые заболевания, не поддающиеся полному излечению и, следовательно, сопровождающие человека на протяжении всей его жизни. Реже встречаются сиротские болезни, ставшие следствием инфекций, аллергических реакций, воздействия плохой экологической обстановки и прочего. Единого мирового стандарта, насколько малораспространенным должно быть заболевание, чтобы считаться орфанным, не существует. Болезнь может быть редкой в одной этнической группе, но достаточно часто встречаться в другой.
Во многих странах критерии орфанности закреплены законодательно. В США редкими признаются болезни, затрагивающие менее 200 тысяч граждан, или примерно одного из 1500 населения, в Японии – менее 50 тысяч, или одного из 2 500 жителей страны. В ЕС, Канаде и Австралии редким считается один случай на 2 000 человек.
Несмотря на статус, общая численность диагностированных орфанных больных существенна: только в ЕС таких пациентов – более 30 млн. Сопоставимую цифру по США приводит американский National Institutes of Health.
В 90‑е годы аналогичные документы были изданы в Сингапуре, Японии и Австралии. В Европе переломным в отношении орфанных препаратов стал 2000‑й год, когда была подписана директива ЕС №141/2000. Несмотря на то что за основу были взяты принципы американского закона, в европейском документе появились свои регламенты: например, в категорию орфанных попадают только ЛС, а не медизделия и товары для диабетиков, что практикуется в США. Срок действия эксклюзивных прав на орфанный препарат в ЕС дольше американского – 10 лет.
Власти США, стремясь сделать терапию доступнее, в 2013 году снизили цены на ряд препаратов, применяемых и в борьбе с орфанными заболеваниями, по программе 340B – для небогатых клиник и пациентов, проходящих по программам Medicare и Medicaid. Однако получилось так, что эта социальная мера пациентов с редкими заболеваниями не коснулась: многие орфанные ЛС предназначены для лечения более распространенных заболеваний, и ценовая протекция затронула именно таких пациентов.
Среди многопрофильных препаратов значится, например, Ритуксан от Roche, который предназначается как для терапии редкого гранулематоза Вегенера, так и для лечения пациентов с банальным ревматоидным артритом, и приобретать препарат со скидкой будут именно последние.
ОРФАННОГО РАДИ
Российская история редких болезней гораздо короче и скромнее мировой. Сам термин «орфанные заболевания» впервые был введен в обращение 1 января 2012 года, в момент вступления в силу закона №323‑ФЗ «Об охране здоровья граждан».
К орфанным отнесли болезни, которые имеют распространенность не более 10 случаев на 100 тысяч человек. Согласно официальной статистике, 300 тысяч россиян страдают орфанными заболеваниями, общий перечень которых состоит из 230 позиций. Однако неофициально называются более удручающие цифры – от 1,5 до 5 млн человек.
Многократный разброс, наверное, можно объяснить – как новизной понятия, так и несовершенством диагностики, скрининга и в целом отсутствием учета пациентов, страдающих редкими болезнями (подробнее – в интервью руководителя Всероссийского общества редких заболеваний Екатерины Захаровой на стр. 29).
Появление термина в федеральном законе позволило правительству издать первый программный документ – постановлением №403 от 26 апреля 2012 года был утвержден подготовленный еще Минздравсоцразвития «Перечень жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни граждан или их инвалидности». В перечень редких заболеваний, поддающихся терапии, вошли 24 нозологии. Но обещанный регулятором федеральный регистр лиц, страдающих этими заболеваниями, до сих пор полноценно не ведется (подробнее – в блиц‑интервью экс‑врио руководителя Росздравнадзора на стр. 18).
Даже на условиях полноценного субсидирования, считают эксперты, децентрализация орфанной программы сразу сделала ее неэффективной.
Осложняет ситуацию и затянувшееся рассмотрение поправок в закон «Об обращении лекарственных средств» №61‑ФЗ: изменения, дающие орфанным препаратам преференции, подготовлены, но пока не приняты.
Впрочем, и принципиальный вопрос – передачи финансирования помощи гражданам, страдающим редкими заболеваниями, с регионального на федеральный уровень – в парламенте наверняка рассмотрят, справедливоросс Андрей Озеров внес в Госдуму соответствующий законопроект.
ДОРОГОЕ НА ДВОИХ
В 2013 году объем лекарственных закупок по нозологиям из «Перечня 24» составил 8,75 млрд рублей. Дистрибьюторов орфанных препаратов не так много – согласно подсчетам, произведенным VM совместно с Headway Company, чуть более полутора сотен. В то же время на ТОП100 приходится 99,9% всех поставок. И лишь два игрока могут назвать совокупную стоимость взятых ими контрактов солидной. На первом месте – компания Александра Апазова «Фармимэкс», собравшая в прошлом году больше половины всей суммы за поставки орфанных ЛС – 4,81 млрд рублей, с минимальной – лишь 0,22% – уступкой от начальной цены. Да и в целом регионам на закупке ЛС от редких болезней сэкономить не удалось, общее снижение начальных цен аукционов составило менее 1% (продолжение на стр. 28).
На втором месте – «Фармстандарт», вовсе не двигавшийся в цене и собравший почти 2,6 млрд рублей. Ровно на эту сумму компания Виктора Харитонина поставила рекомбинантный фактор свертывания Коагил‑VII (МНН: эптаког альфа [активированный]). Вклад других дистрибьюторов в поставки этого ЛС незначителен – примерно 110 млн рублей. Весомое участие «Фармстандарта» в аукционах по поставке Коагила‑VII не случайно: выпускает препарат его стопроцентная «дочка» – ЗАО «Генериум». По тому же МНН был закуплен еще один препарат – Новосэвен от Novo Nordisk, но по сравнению с Коагилом‑VII стоимость этой поставки была в 20 раз скромнее – 242 млн рублей. На двоих «Фармимэкс» и «Фармстандарт» выиграли контракты на общую сумму в 7,41 млрд рублей, забрав 85% совокупной стоимости всех разыгранных лотов. Остальные восемь дистрибьюторов из ТОП10 суммарными усилиями не добрались и до отметки в 1 млрд рублей. Если посмотреть на карту поставок, то и здесь безусловным лидером выступает «Фармимэкс», сохранивший за собой первенство в 39 из 74 проводивших торги регионов. На второй позиции – «Р‑Фарм», обогнавший соперников в восьми регионах и в целом выручивший на орфанных ЛС около 160 млн рублей. «Фармимэкс» лидирует и по линии МНН, которых в нынешнем орфанном списке всего 15. Не участвовала компания в аукционе по закупке препаратов с МНН: эптаког альфа [активированный] – по понятным причинам и еще в нескольких из‑за незначительности ценового веса лотов. У большинства закупавшихся в 2013 году орфанных препаратов нет аналогов, поэтому многим МНН соответствовало лишь по одному торговому наименованию. Помимо уже упомянутого МНН: эптаког альфа [активированный] с Коагил‑VII и Новосэвен, в ряду исключений – кальцитонин. Это МНН представляют четыре препарата – Вепрена (ООО «Натива»), Алостин (Apotex), Миакальцик (Novartis Pharma) и Остеовер (ОАО «Верофарм»). По объему закупок за МНН: эптаког альфа [активированный] следует МНН: идурсульфаза (Элапраза от Shire Human Genetic Therapies Inc) с показателем 1,59 млрд рублей. Третью позицию с 1,49 млрд рублей занимает МНН: экулизумаб (Солирис от Alexion Pharma). За ним с небольшим отрывом следует МНН: имиглюцераза (Церезим от Genzyme), собравшее 1,34 млрд рублей. Объемы закупок препаратов с остальными МНН ощутимо меньше. Список производителей закупленных орфанных препаратов состоит всего из 11 позиций. Три из них занимают российские компании – «Натива», «Верофарм» и, конечно, «Генериум», благодаря внушительному вкладу которого доля отечественных препаратов в совокупной стоимости закупок составляет почти треть.
«ПЕРЕЧЕНЬ 24» – ЗАСЛУГА ТАТЬЯНЫ ГОЛИКОВОЙ»
Как начинался орфанный госпроект
Текст: Ольга Макаркина
Бывший врио руководителя Росздравнадзора Елена Тельнова объяснила VM, почему пациенты, страдающие редкими заболеваниями, не получают гарантированную государством помощь в полном объеме.
– Программа рождалась на ваших глазах, как вы оцениваете ее эффективность?
– По сути, программы никакой нет, и о ее эффективности судить пока еще рано. Однако в этом направлении за последнее время есть значительные подвижки. Главная же проблема в том, что лекарственным обеспечением терапии орфанных заболеваний, к сожалению, занимаются исключительно регионы, при этом никакого дополнительного финансирования им никто на это не выделил. И на эту совсем не дешевую помощь у многих субъектов денег нет.
– Так как же, по идее, может работать система поддержки таких людей?
– Перечень из 24 редких заболеваний – всего лишь список. На мой взгляд, основополагающим должен был стать регистр пациентов, страдающих орфанными заболеваниями: необходимо понимание числа больных, их состояния, наличия или отсутствия эффективной терапии и зарегистрированных препаратов. Когда мы замышляли программу «Семь нозологий», мы начали с реестра пациентов, нуждающихся в дорогостоящих препаратах. Причем один реестр мы самостоятельно составили по оценкам региональных министерств, а другой – с помощью западных компаний, знающих аудиторию своих пациентов. Наложив эти перечни, мы посмотрели, насколько они совпадают и разнятся, произвели подсчеты – так была основана система госпрограммы, которая сегодня эффективно работает.
«Перечень 24» формировался по схожему принципу, но развития программа не получила. До сих пор Минздрав, обещавший вот‑вот разработать порядок составления регистра пациентов, ничего не сделал. Многие орфанные заболевания лечатся симптоматически, по большинству из них, к сожалению, вообще нет препаратов. И подход по‑прежнему остается формальным – передали проблемный сегмент гособязательств субъектам, а те пусть сами разбираются. И никто толком не посчитал, а может быть, федеральный центр вполне способен взять на себя финансирование наиболее затратной части орфанной госпрограммы. Понятно, что сегодня с бюджетом у нас не все так хорошо, как было раньше. Но очевидно, что какие‑то вопросы, именно ради эффективности расходов, нужно решать на федеральном уровне.
– Но ведь по ряду орфанных заболеваний есть эффективные зарубежные препараты, перспективные разработки, не получившие пока производственных лицензий, или лекарства, не зарегистрированные в России. Почему эти лончи у нас не проходят?
– Невыгодно. Орфанные препараты – дорогие, и потребность в них не очень большая, при этом нет уверенности в стабильных поставках. И если бы фармацевтические компании знали, что закупка их препарата будет гарантирована в определенном объеме, то, наверное, у них бы и отношение к нашему рынку было другим. А кто будет регистрировать препарат в России, если один регион просит закупить препарат для одного или двух больных, а другой регион вообще игнорирует проблему – по бедности или недосмотру. А что завтра будет, вообще неизвестно.
Да, существует возможность приобретения незарегистрированных препаратов [порядок ввоза незарегистрированных ЛС определен постановлением правительства от 29 сентября 2010 года № 771. – VM]. Но это единичные случаи. Либо препарат приобретает и ввозит благотворительный фонд, либо лекарство предоставляет компания‑производитель, либо клиника закупает его для своих пациентов. Для такой неритмичной, случайной поставки компания не станет тратиться на регистрацию препарата в России, проще предоставить по запросу незарегистрированный.
Повторю, необходима стабильность в закупках. Я говорю про централизованную закупку, и необязательно через Минздрав. Регулятор должен выполнять свою основную функцию: формулировать политику охраны здоровья граждан, вырабатывать нормативную базу, выстраивать действенную систему медицинской и лекарственной помощи, разрабатывать и утверждать технологии этих процессов, а не скатываться к выполнению роли закупочного агентства.
– То есть в регионы орфанную программу нельзя было отдавать даже на условиях полноценного субсидирования?
– Объясню, почему нельзя. Как правило, число больных и, как следствие, потребность в препаратах – небольшие. Размывать этот бюджет и функционал по территориям невыгодно. Да, регион получит деньги, но не факт, что закупит необходимое, хотя деньги могут поступить целевым назначением. Я говорю не про все регионы. К сожалению, в ряде регионов такая ситуация наблюдается.
– Так как же обеспечить фармакотерапию пациентов с орфанными заболеваниями?
– Определить четко потребность, составить регистр, определить объемы закупок и отдать все это в федеральный центр. Включить определение орфанных заболеваний в закон – мало, хотя, конечно, очень важно. Причем это было сделано еще при прежнем министре – Татьяне Голиковой. Как и «Перечень 24» – ее заслуга. Необходимо двигаться дальше.
«Оптимисты». Как живут люди с орфанными заболеваниями в России?
Ника Воюцкая
Павлику, сыну Снежаны Митиной, было всего три года, когда ему поставили диагноз мукополисахаридоз II типа, или синдром Хантера. «Мы только открыли дверь, а врач уже говорит: „Какой прекрасный Хантер!“ — рассказывает Снежана теперь. — С первой частью я согласилась, а вот кто такой Хантер, мне тогда было неизвестно».
В детском саду мальчик был самым высоким в своей группе, читал стихи про бычка, который идет и качается. Поверить, что совсем скоро он перестанет ходить и говорить, казалось невозможным.
Но в четыре из-за гиперактивности Павлика исключат из детского сада, в шесть — из детского сада для инвалидов: перестанет усваивать программу. У него изменятся внешность и характер.
Терапии, способной корректировать часть симптомов, вместе с другими родителями Снежане придется добиваться самой. Элапразу — единственный препарат, способный помочь ее сыну, зарегистрируют в России не сразу и именно ее трудами.
Синдром Хантера — это одна из форм мукополисахаридоза, генетическое заболевание, возникающее в результате дефицита ряда ферментов, расщепляющих продукты обмена. Этот дефицит приводит к накоплению белково‑углеводных комплексов и жиров в клетках. В результате чего происходит «самоотравление» организма и поражаются органы.
В России редким, или орфанным, считается заболевание, которое встречается у 1 человека из 10 тысяч или реже. Именно из-за редкости такие болезни сложно диагностировать и лечить. Тот синдром, что у Паши, встречается у одного ребенка на 132 000 родившихся младенцев.
Сейчас Снежана помогает больным мукополисахаридозом как президент МБОО «Хантер-синдром»: рассказывает, как получить лекарство, через благотворительные фонды находит памперсы, инвалидные коляски и ортопедическую обувь.
Право выбора
Большинство редких болезней хронические. То есть неизлечимые. Почти все приводят к инвалидизации и смерти. Орфанные препараты, если терапия от конкретного недуга все-таки существует, как правило, не излечивают болезнь полностью, а лишь снимают тяжесть симптомов и приниматься должны на протяжении всей жизни.
«С первой частью я согласилась, а вот кто такой Хантер, мне тогда было неизвестно»
Лечение двадцати четырех самых тяжелых и дорогих орфанных заболеваний финансируется государством. Если диагноз входит в знаменитый список «12 нозологий», то лекарство закупают за счет федерального бюджета, если в «Перечень 24» — за препараты платят регионы. В остальных случаях пациенты получают терапию как жизненно необходимую или по инвалидности.
Известно, что крайне важно начать лечение от мукополисахаридоза как можно раньше. Павлик получил первые свои медикаменты только в восемь. Сейчас ему девятнадцать, с восемнадцати он не говорит, хотя понимает человеческую речь.
«Конечно, мне часто пишут мамы, — признается Снежана: „Мы видели вашего ребенка в интернете. Он не разговаривает и ходит в памперсах. Зачем нужна такая жизнь?“. Но у меня короткий ответ. Что вы выберете: двенадцать лет ходить на кладбище или двенадцать лет целовать сына?».
За двенадцать лет терапии Павел не пропустил ни одного приема Элапразы. Хоть препарат и стоит около миллиона (!) рублей в неделю. Благо в Москве проблем с лечением нет.
Вопрос денег
У сына Натальи Буртаевой мукополисахаридоз IV типа, заболевание схожее, но ни в списке «12 нозологий», ни в «Перечне 24» его нет.
Даниилу уже шестнадцать. Его рост 97 сантиметров. Ходит с трудом: ноги и руки деформированы, внутренние органы тоже деформированы. С пятого класса Даня учится на дому. Нервная система при этом диагнозе не страдает, дети с ним отлично осваивают школьную программу. Но поврежденной оказывается опорно-двигательная система, страдает слух.
Препараты специальной ферментной заместительной терапии, положенной в таких случаях, существуют, но первое время были не зарегистрированы в России. Единственное лекарство, одобренное американским надзорным фармакологическим ведомством (FDA) в 2014 году, называется элосульфаза альфа — торговое название Вимизим (Vimizim).
Без Вимизима больные мукополисахаридозом IV типа умирают к двадцати годам. Когда новости о его разработке только появились в специализированных СМИ, Наталья было обрадовалась. Но стоимость его годового курса превышает 60 миллионов рублей.
Минздрав Ульяновской области ожидаемо отказался приобретать препарат. Наталья подала в суд и выиграла: первый в 2016 году, второй, когда министерство подало апелляцию, — в 2017. Ни письма в Росздравнадзор и Путину, ни сюжет на «Первом канале» не помогли: Даниил до сих пор не получает лечения.
«Мы не прячемся, — твердо сообщает Наталья корреспонденту. — Даня часто гуляет на коляске. Мы с ним ездили на море, а сегодня вообще особенный день: последний звонок. Он выйдет на сцену и получит аттестат зрелости.
Добавляет: «Он у меня обычно говорит, когда я переживаю, что люди его не примут: „Мам, ты, это, не переживай. Я привык, что на меня так смотрят“».
Более того, после школы Даниил планирует учиться дальше, признается мать, вздыхая: «Не знаю, сколько он будет жить». Врачи точно ответить на этот вопрос тоже не в силах, но очевидно, что без терапии Даня проживет недолго.
«Сделано в России»
Почему орфанные препараты стоят так дорого? Фармакологические компании разрабатывают их ради прибыли. Исследования нового лекарства стоят немало, а весьма небольшой тираж препарата призван не только возместить потраченные на него миллионы, но и «вернуться» с процентами.
Если в производстве инсулина в России заинтересованы миллионы, то «целевая аудитория» орфанных препаратов даже на такую большую страну, как наша, нередко не превышает пары десятков пациентов.
Во многих странах предусмотрена государственная поддержка исследователей, разработчиков и производителей орфанных препаратов: льготы, гранты и так далее. Но в России таких программ нет.
«Мам, ты, это, не переживай. Я привык, что на меня так смотрят»
Для того чтобы снизить издержки, власти предпочитают не субсидировать или договариваться с производителем, а искать оригинальным препаратам дешевые аналоги, дженерики. К сожалению, как убеждены некоторые эксперты, они нередко отличаются по эффективности.
Насте Катасоновой двадцать один, и она не знает, сколько раз лежала в больнице: до восьми лет — раз в год, до четырнадцати — по два раза, потом — по три, сейчас — госпитализироваться приходится чуть ли не каждый месяц.
Говорит, что в больнице ей спокойно: за твоим состоянием следят специалисты. Впрочем, и тут есть поводы для волнений. У Насти муковисцидоз, редкая болезнь легких, и недавно оригинальные антибиотики заменили отечественным дженериком.
Внешне люди с редкими заболеваниями часто не отличаются от здоровых. Однажды в школе кто-то из родителей одноклассников сказал, что девочке родственники купили инвалидность, дабы та не ходила на уроки. Одноклассники в ответ на слух объявили несчастной бойкот. Теперь она о муковисцидозе говорит открыто, чтобы не возникало кривотолков.
В ноябре Насте двадцать два. Между больницами и процедурами она профессионально фотографирует, ведет личный блог, помогает с соцсетями благотворительным организациям, занимается веганским магазином.
От побочных эффектов нового дженерика — у оригинального препарата их не было — состояние Анастасии ухудшилось. «Но не пить их нельзя, иначе — захлебнешься в мокроте», — объясняет пациентка.
«Каждое утро для меня — испытание, — рассказывает она. — Мокрота за ночь отлеживается в легких. Встаю. Но не так красиво, как в фильмах, а с кашлем. За день получается стакан густой зеленой жижи. Таблетки, ингаляция, если надо на работу, приходится просыпаться за два часа до выхода». Днем опять ингаляции. Перед сном питание: через гастростому. Это специальную трубка, которая устанавливается в отверстие на животе и ведет прямо в желудок.
Екатерина Захарова, руководитель лаборатории наследственных болезней обмена веществ, председатель экспертного совета по редким болезням Всероссийского общества орфанных заболеваний, считает, что нужен активный диалог с разработчиками и производителями орфанных препаратов: «Необходимо выяснять, какие лекарства фармкомпании могут предложить бесплатно, какую минимальную стоимость могут установить. Кроме того, важно понять, какие препараты Россия может производить самостоятельно, чтобы обеспечить своих граждан: возможно, в рамках госзадания будет дешевле создать российское лекарство для небольшого числа пациентов».
Преступная забывчивость
Впрочем, не все орфанные препараты стоят миллионы. Вот только получить даже относительно недорогое лекарство нуждающимся в нем порой оказывается совсем не просто.
У Виктории Рыжковой редкое генетическое заболевание. Синдром Вильсона-Коновалова, или гепатоцеребральная дистрофия.
Медь при этой болезни не выводится из организма, а проблемы с нарушением ее обмена ведут к накоплению элемента в нервной системе, почечной, печеночной тканях и роговице, что оборачивается токсическим повреждением указанных органов.
Что такое орфанный препарат
Добродетельная цель обеспечения фундаментального равенства и неотъемлемости права на поддержание здоровья людей, страдающих от заболеваний с низкой (относительно других) распространённостью исходит из духа Всеобщей декларации прав человека, принятой Организацией Объединённых Наций в 1948 г. [1].
История специальных мер и принципов защиты пациентов с редкими заболеваниями и поощрительных мер для разработчиков соответствующих лекарственных средств (ЛС) началась в Соединённых Штатах Америки. Созданное там в 1906 г. Управление по контролю пищевых продуктов и лекарственных препаратов США (FDA) устанавливает нормы в принципах регулирования области обращения ЛС и непосредственно контролирует соблюдение соответствующих законов. Однако до 1938 г. серьёзные обеспечительные рычаги влияния на безопасность и эффективность поступающих в США в продажу ЛС у FDA отсутствовали.
В 1938 г. в США был принят Федеральный закон о пищевых продуктах, лекарственных и косметических средствах (англ. Federal Food, Drug, and Cosmetic Act): он был подписан Президентом Франклином Рузвельтом 25 июня 1938 г. [2]. Принятию закона содействовали общественный резонанс, обращения к Президенту США и результаты исследований FDA в отношении причин смерти более 100 пациентов в 15 штатах, принявших ядовитую суспензию стрептоцида (Elixir sulfanilamide), которую фирма С.Э. Массэнджилл (S.E. Massengill Company) выпустила в 1937 г. на рынок без каких-либо предварительных исследований безопасности; антибактериальный препарат растворяли в смеси, содержащей диэтиленгликоль (Диэтиленгликоль токсичен: при попадании в организм вызывает острое отравление, поражает почки и печень (ГОСТ СССР «10136-77 – Диэтиленгликоль» // ОКП 24 2213 0000»), однако до инцидента с «Elixir sulfanilamide» это было неизвестно) с целью получить жидкую лекарственную форму [3]. До принятия этого Закона лекарственные препараты в США могли выпускаться в свободную продажу без ограничений, по желанию разработчиков или производителей [4]. Так впервые было принято законодательное требование доказывать безопасность препаратов перед разрешением их широкого использования. Закон о пищевых продуктах, лекарственных и косметических средствах действует в США и по настоящее время, и все изменения регуляторных и защищающих пациентов законодательных требований вводятся поправками к этому основному закону о ЛС.
Уже к 1960-м гг. возросшие требования к качеству, эффективности и безопасности ЛС, во многом обусловленные ещё одной трагедией – массовым бездоказательным применением препаратов талидомида беременными женщинами в Европе и Канаде (В США благодаря принципиальности и научной объективности сотрудницы FDA Фрэнсис Келси (Frances Oldham Kelsey; 1914-2015) талидомид, заявленный компанией «Ричардсон Меррелл» (Richardson Merrell) под торговым названием Kevadon, так и не получил одобрения регулятора и не поступил в национальное обращение (в 1962 г. Ф. Келси получила за это Президентскую награду за выдающуюся гражданскую службу).), обусловили принятие законопроекта (одобрен 87-м Созывом Конгресса США, подписан Президентом Джоном Кеннеди 10 октября 1962 г.), известного как «Поправка Кефовера-Харриса» (Kefauver Harris Amendment), или Поправка об эффективности лекарственных средств [5, 6]. Эта поправка к Закону о пищевых продуктах, лекарственных и косметических средствах обязала производителей ЛС представлять при государственной регистрации доказательства их эффективности (до этого требовалось доказывать только их безопасность), указывать в рекламе ЛС и в их инструкциях по применению достоверную информацию о побочных реакциях, а также в определенной степени регулировала вопросы производства воспроизведенных препаратов. Впервые в мире было введено требование информированного согласия пациента на участие в клиническом исследовании (КИ), что положило начало эре контролируемых КИ и введению принципов GCP (надлежащей клинической практики) при проведении таких исследований, включая подачу точных и своевременных сведений о нежелательных явлениях. Фармацевтические компании уже не могли делать рекламных заявлений об эффективности своих препаратов, которые не были бы подкреплены научными доказательствами и не были бы включены в инструкцию по применению (текст проекта которой подлежал бы научной экспертизе в FDA) [6, 7].
Поправка Кефовера-Харриса и вызванные ею последующие ужесточения регистрационных и регуляторных требований полностью изменили систему одобрения и государственного контроля над обращением ЛС в США, а вслед за ними и в остальных странах [8], оказала решающее влияние на возникновение современных принципов экспертизы ЛС для медицинского применения, центральным из которых является строго положительное отношение ожидаемой пользы медицинского применения лекарственного препарата к возможному (и в приемлемой степени максимально изученному до одобрения) риску его применения. В то же время стоимость научно-исследовательских работ, результатами которых разработчики новых препаратов должны были обосновать экспертному учреждению целесообразность одобрения своего продукта, многократно возросла, и, как следствие, в новых условиях фармацевтические компании стали концентрировать свои ресурсы на разработке препаратов с потенциально большой целевой группой пациентов как потребителей создаваемого ЛС для возмещения всё возрастающих расходов на исследования. Неизбежно с этим более редкие заболевания, от которых страдали тысячи и десятки тысяч – но не миллионы – людей стали игнорироваться разработчиками, поскольку экономическая выгода от продвижения соответствующих лекарственных и диагностических средств до уровня национального оборота была низкой или несущественной, и приобрели название «осиротевших» (орфанных, от англ. orphan – сирота). За 10-15 лет после принятия поправки Кефовера-Харриса создалась ситуация, когда новых эффективных препаратов для лечения редких заболеваний просто не создавалось, или количество таких препаратов было очень мало: примерами таких редких (для США) заболеваний являются лепра, болезнь Хантингтона, муковисцидоз, боковой амиотрофический склероз, некоторые аритмогенные кардиомиопатии и др. [9, 10].
К 1983 г. в США было одобрено только 38 препаратов для лечения редких заболеваний [11]. В начале 1980-х гг. Эби Мейерс (Abbey Meyers), чей сын страдал от синдрома Туретта, вместе с пациентами с орфанными заболеваниями и членами их семей, представителями благотворительных движений создала Национальную Организацию редких заболеваний (National Organization for Rare Disorders, NORD), и стала её президентом [12]. Пациентов с синдромом Туретта в США было приблизительно 100 000, и поэтому сын Эби, получая экспериментальное медикаментозное лечение, тем не менее не мог рассчитывать, что разработчики и производители ЛС будут экстенсивно развивать неприбыльные направления и экономически неперспективные исследования.
В свете стремления улучшить общее благополучие жителей Планеты, присущего 1970-м гг., и повышения внимания к вопросам здоровья, Национальная Организация редких заболеваний (ещё до своего официального оформления), и другие активисты лоббировали принятие Закона об орфанных лекарственных препаратах (Orphan Drug Act) как поправки к Федеральному закону о пищевых продуктах, лекарственных и косметических средствах (Закон о внесении изменений в Федеральный закон о пищевых продуктах, лекарственных и косметических средствах для способствования разработкам лекарственных препаратов для редких заболеваний и расстройств, и для других целей) [13]. Главным формальным инициатором этого законопроекта (H.R. 5238) был Генри Ваксман (Henry Waxman), председатель подкомитета по здоровью (в составе комитета Палаты представителей по энергетике и коммерции). 4 января 1983 г. Президент Рональд Рейган подписал законопроект H.R. 5238 и обратился к согражданам со специальным обращением, посвященным этому акту, которое заключил словами: «Жаль только, что одним росчерком пера я не могу также постановить, чтобы прекратились боль и муки людей, страдающих от этих болезней» («I only wish with the stroke of this pen I could also decree that the pain and heartache of people who suffer from these diseases would cease» [10]).
Закон устанавливал главный критерий, по которому лекарственным препаратам, вакцинам и диагностическим средствам мог быть присвоен орфанный статус: это предназначение для лечения (или диагностики) заболевания, которое затрагивало менее 200 000 граждан США. Такой статус давал компании-разработчику данного средства право исключительной (монопольной) его реализации в течение 7 лет (Англ. «market exclusivity (period)»: не зависит от патентного статуса препарата, более того – его срок отсчитывается только с момента одобрения регулятором данного препарата. При этом если в течение действия данного исключительного права другая компания разработала препарат для лечения этой же редкой болезни, то она должна доказывать, что её препарат явно фармакологически превосходит (например, он менее токсичен или более эффективен), чем уже одобренный по данному показанию препарат (так, препарат Rebif (разработчик Merck Serono, Германия) 07.03.2002 г. «ниспроверг» исключительное рыночное право препарата Avonex (разработчик Biogen, США) в силу своего клинического превосходства (МНН – интерферон бета 1а)). Таким образом, «исключительное рыночное право» отличается от простой патентной защиты новой молекулы и является наиболее весомым (с позиции производителей) преимуществом среди мер правительственной поддержки орфанных препаратов), ускоренную процедуру рассмотрения препарата, налоговый кредит до 50 % стоимости разработки, и другие поощрения.
Эффективность декларированных Законом об орфанных препаратах стимулирующих мер явствует из следующих цифр. С момента его подписания к маю 2010 г. FDA одобрило 353 орфанных препарата и предоставило орфанный статус 2 116 молекулам и соединениям [14], к февралю 2015 г. было одобрено уже 511 орфанных препаратов, орфанный статус был предоставлен в 3 280 случаях (при этом общее число заявок в FDA на такой статус составило около 4 700) [15]. Более 200 орфанных заболеваний стали рассматриваться в медицине как излечимые или медикаментозно купируемые [14]. Так, например, медианный возраст выживаемости при муковисцидозе в США в конце 1950-х гг. составлял 6 месяцев; в 2010 г. этот показатель составлял около 37 лет для женщин и 40 лет для мужчин [16] (при этом муковисцидоз диагностируется с одинаковой частотой у мужчин и женщин).
Орфанные ЛС ныне являются более чем значительной областью всей фармацевтической разработки. Например, в 2014 г. из 41 нового (оригинального) препарата для медицинского применения (не включая вакцины, клеточные, тканевые и генотерапевтические средства), одобренного в США, как сообщается в ежегодном отчёте Центра по экспертизе и исследованиям ЛС при FDA (Center for Drug Evaluation and Research), 17 были препаратами для лечения орфанных заболеваний (41 %). Это Cerdelga (eliglustat) для лечения болезни Гоше́ типа 1, Vimizim (elosulfase alfa) для лечения мукополисахаридоза IVA типа, Sylvant (siltuximab) для терапии многоочаговой болезни Кастлемена и другие инновационные препараты [17]. Более того, в 2015 г. уже 21 из 45 (то есть 47 %!) одобренных в США оригинальных ЛС были зарегистрированы в статусе орфанных, включая Orkambi (lumacaftor/ivacaftor) для пациентов от 12 лет с муковисцидозом [18].
В современном мире, основанном на информации, её свободном потоке и постиндустриальной глобальной сетевой экономике, финансовый капитал вообще перестал быть критически определяющим фактором, как это было все последние пять столетий – с расцвета свободной коммерции, порождённого Великими географическими открытиями [25]. В информационную эпоху, когда человек фактически не привязан в рамках профессионального и вообще онтологического выбора к пространству, паттернам предыдущих поколений, стоимость средств производства в большинстве случаев пренебрежимо мала, доступ к освоенным человечеством знаниям мгновенен, а компании озабочены не поиском денег, а поиском идей, куда их есть смысл инвестировать – капитал не является ныне ограничивающим ресурсом. Сегодня, как никогда прежде в истории, все достижения определяются именно самим человеком – его способностью развиваться, а главным вопросом прогресса становится безграничный потенциал этой способности.
Часть I. Редкие (орфанные) заболевания
Редкие заболевания отличаются большим видовым разнообразием: по оценкам, их более 7 000, каждую неделю в медицинской периодике появляются сообщения о новых выявленных заболеваниях. Частота их встречаемости варьирует: так, недостаточность фосфопентозоизомеразы (рибозо-5-фосфат-изомеразы, КФ 5.3.1.6), фермента пентозофосфатного пути катаболизма глюкозы, катализирующего превращение D-рибулозо-5-фосфата в его альдоизомер, D-рибозо-5-фосфат, вызываемая мутацией RPI(Ala61Val) в гене данного белка (причём экспрессия мутантного аллеля различается в разных тканях), была обнаружена в 1999 г. у юноши 1984 г. рождения, и более пациентов с такой энзимопатией диагностировано пока не было [26]. Эта болезнь, таким образом, является самой редкой на Земле. Патология клинически проявляется как прогрессирующая лейкоэнцефалопатия (дисплазия substantia alba), а также периферической нейропатией и нарушениями метаболизма полиолов [27]. Другое редкое заболевание, наследственная оротацидурия, передающееся по аутосомно-рецессивному механизму, характеризуется недостаточностью сразу двух ферментов: оротат-фосфорибозилтрансферазы (КФ 2.4.2.10) и оротидин-5’-фосфат-декарбоксилазы (КФ 4.1.1.23) [28]. Патология проявляется в виде анемии, лейкопении, агранулоцитоза, отставанием в развитии и др., и известно только приблизительно 20 человек в мире, страдающих от этой болезни [29].
Другие редкие заболевания могут быть не столь редкими (десятки и сотни тысяч страдающих), однако, подобно этим двум патологиям, большая часть их является врождёнными, обусловленными мутациями (главным образом генными). Как правило, они также характеризуются трудностью и длительностью диагностирования (чем реже болезнь, тем соответственно меньше врачей обладают опытом её распознавания и лечения), тяжестью (так как правильная диагностика зачастую занимает несколько месяцев и даже лет) и низким качеством жизни; в ряде случаев редкие болезни неизлечимы на данном этапе развития человечества. Этиология их редко бывает ясна, спекулятивны также патогенетические механизмы. Это во многом обусловлено малым (относительно других заболеваний) количеством исследований, хотя некоторые редкие заболевания интенсивно изучаются (муковисцидоз, болезнь Фабри, гемофилия и др.), либо были достаточно хорошо изучены предыдущими поколениями (нарушения обмена ароматических аминокислот, лепра).
Особые черты большинства редких заболеваний отражаются и в сложности разработки препаратов для их лечении – орфанных препаратов. Подавляющее большинство их – это моноклональные антитела, иные белковые молекулы (например, различные компоненты гемокоагулянтной системы), в ряде случаев синтетические фрагменты нуклеиновых кислот, антисмысловые олигонуклеотиды [9, 15, 17, 18]. В настоящее время бóльшая часть орфанных препаратов представляет собой средства для патогенетической либо заместительной терапии, однако большие надежды возлагаются на средства передовой терапии, такие как, например, клеточные трансплантаты в органы с нарушенной функцией или генотерапевтические препараты, в том числе для фетальной генотерапии.
Вместе с тем, не все препараты для лечения редких заболеваний являются сложными белковыми молекулами или инновационными высокомолекулярными соединениями: так, одобренный FDA 04 сентября 2015 г. первый препарат для лечения наследственной оротацидурии Xuriden представляет собой достаточно простую с точки зрения биохимии молекулу – нуклеозид, уридина триацетат [29]. Xuriden (инноватор – Wellstat Therapeutics Corporation, Gaithersburg, Мэрилэнд, США) предназначен для применения per os, и является средством заместительной терапии, возмещающим в организме недостаток уридина.
Следует особо подчеркнуть, что редкие заболевания могут быть редкими в одной части света и распространёнными в другой; частота заболевания также изменяется во времени. Например, лепра (проказа), которая в Античности и во времена Европейского Средневековья являлась распространённым, социально значимым заболеванием, ныне в России и США является очень редким (несколько сотен пациентов) заболеванием, однако в ряде глубинных районов Индии, Бангладеша и Бразилии по-прежнему распространена. Чёрная оспа, которая в предыдущие столетия была угрозой общемирового масштаба, жесточайшим ужасом целых континентов, в XVIII веке была существенно ограничена в распространении благодаря научным достижениям, ставшим возможными в эпоху светского Просвещения, а в 1958-1980 гг. по инициативе врачей-активистов Советского Союза и вовсе уничтожена на Планете [30]. Однако поднимаются новые заболевания, которые из неизвестных ранее или малораспространённых могут стать или уже стали по разным причинам угрозами международного характера (лучевая болезнь, ВИЧ-инфекция, алиментарные дислипидемии и др.), либо которые из эндемических заболеваний в силу глобализации имеют тенденцию стать таковыми (легионеллёз, геморрагическая лихорадка Эбола, лихорадка Зика и др.).
Вместе с тем, в любом случае для большого числа болезней так или иначе свойственны национальные (связанные с происхождением индивидуума), эндемические или социальные особенности, влияющие на их эпидемиологию. Так, например, муковисцидоз (эта патология относительно хорошо изучена, и в отношении её терапии достигнуты, как уже упоминалось, значительные (сравнительно с наследственными заболеваниями в целом) успехи) является весьма распространённой болезнью среди представителей Европеоидной расы (частота встречаемости 1:3300). Среди жителей стран Латинской Америки он встречается с частотой 1:9500. Однако в тоже время среди представителей Монголоидной и Негроидной рас (проживающих в географически коренных регионах) муковисцидоз существенно редок: частота его менее 1:50000. Но среди этнических представителей этих рас, проживающих в США, частота данной патологии уже выше: 1:15300 для Негроидов и 1:32100 для Монголоидов) [31], что скорее всего объясняется метисацией. А самая высокая доля этого аутосомно-рецессивного заболевания зафиксирована в Ирландии: по состоянию на 2000 г. муковисцидоз диагностировали там у одного из 1 353 человек [32].
Этот пример демонстрирует, что «орфанность» заболевания не может просто экстраполироваться на основании регуляторного опыта или эпидемиологической картины в других странах. Критерий «орфанности», по крайней мере для крупных государств, должен устанавливаться на национальном уровне, и при этом в дальнейшем он может быть пересмотрен по результатам эпидемиологического мониторинга. В России, как в многонациональном и громадном по территориальной протяжённости государстве, частота встречаемости муковисцидоза аналогичным образом имеет и значительные региональные различия. По данным исследований 2007-2011 гг., доложенным на Х Национальном конгрессе «Муковисцидоз у детей и взрослых» (г. Ярославль, 1-2 июня 2011 г.) она колеблется от 1:2500 до 1:17000, а в среднем по Федерации составляет 1:10000 [33]. То есть в абсолютном выражении это приблизительно 15 000 пациентов (для сравнения, по состоянию на 2013 г. в США – 28 103 человек, а всего в мире – около 70 000 [34].
С целью повышения информированности общества и органов государственной власти о редких заболеваниях и проблемах пациентов и в знак солидарности с ними, с 2008 г. (в России – с 2009 г.) по инициативе некоммерческой организации «Европейская организация редких заболеваний» (European Organization for Rare Diseases, EURORDIS), в последний день февраля отмечается День редких заболеваний [35]. В 2016 г. этот день был отмечен 29 февраля [36].
Часть II. Международный опыт
США стали первым государством, законодательно внедрившим национальную систему поддержки разработки орфанных препаратов. Сравнительные исследования показывают, что США обладают наиболее развитыми системой регулирования и нормативно-правовой базой в сфере обращения ЛС [37], поэтому законодательные инициативы этой страны оказывают существенное влияние на другие.
Вслед за США орфанное законодательство (ОЗ) приняла Япония, которая является одной из стран с наиболее эффективной системой общественного здравоохранения в мире: правительство ежегодно тратит более 300 млрд USD на расходы в этой области [38]. Уже в 1985 г. в Японии было постановлено, что для препаратов для лечения редких заболеваний объём регистрационного досье может быть уменьшен, а его рассмотрение будет проводиться в ускоренном режиме (ускоренная процедура регистрации). В 1991 г. ОЗ было принято в Республике Сингапур (население около 5,5 млн человек), в апреле 1993 г. – в Японии – уже как соакцентированный пересмотр Закона о фармацевтической деятельности. Японское право в данной области в целом реплицирует соответствующее законодательство США, но при этом поощрительные меры там также распространяются на медицинские изделия (Что имеет следствием опережающее развитие медицинских технологий и доведение их до пациентов. Так, 30.01.2016 г. Минздрав Японии впервые включил использование киборг-костюмов, которые помогают двигаться и осуществлять многие волевые физические действия пациентам со спинно-мускульной атрофией и боковым амиотрофическим склерозом, в систему обслуживания по медицинскому страхованию (таких пациентов в Японии насчитывается приблизительно 3 400 человек при населении 127 млн) [39]) [40].
В Австралии ОЗ было введено в 1998 г., в Европейском Союзе – с 2000 г. (Постановление (EC) № 141/2000) [41]. Как правило, ОЗ не имплементируется, а принимается в государстве с учётом местных правовых, экономических и иных особенностей. В то же время 08.11.2007 г. FDA и Европейское агентство по лекарственным средствам (ЕМА) объявили, что отныне будут использовать единую форму заявления о предоставлении лекарственному препарату орфанного статуса (англ. Common Application for Orphan Medicinal Product Designation) с целью уменьшения административного бремени для тех компаний, который намерены регистрировать свой инновационный препарат и в США, и в Евросоюзе, и тем самым повышения доступности орфанных ЛС на территории обеих юрисдикций [42].
Данные по особенностям регистрации и регулирования обращения орфанных ЛС в некоторых ведущих странах суммированы в таблице.
Особенности орфанного регулирования в разных государствах и союзах



